De novo Assembly followed by read mapping to the assembly using several mappers
- Author
Menachem Sklarz
- Affiliation
Bioinformatics Core Facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
This workflow demonstrates the use of the index building and mapping modules.
It creates an assembly and maps the reads to it.
Steps:
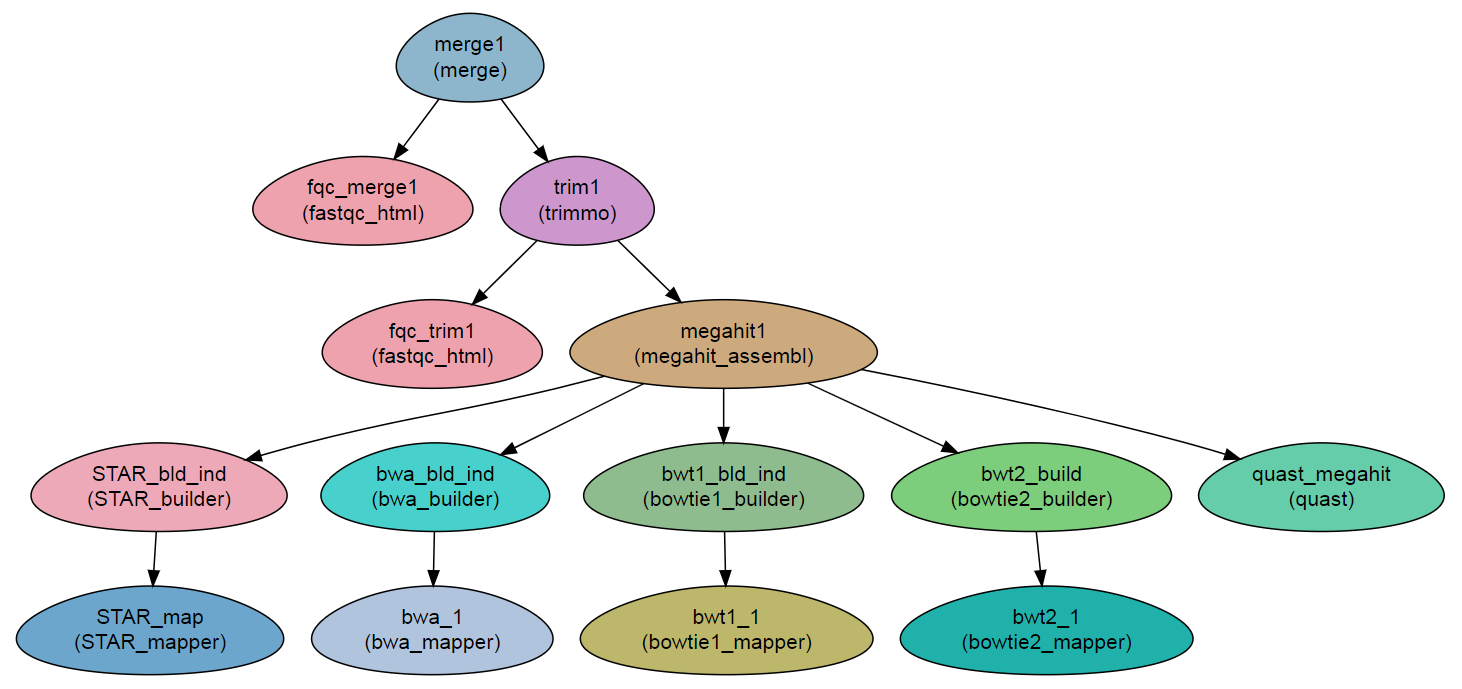
Merging (
merge), quality testing (fastqc_html) and trimming (trimmo).A sample-wise assembly is constructed with
megahit.Quality testing of the assembly with
quast.An index for the assembly is created with
bowtie2_builder,bowtie_builder,bwa_builderandSTAR_buildermodules.The reads are mapped to the assembly with
bowtie2_mapper,bowtie_mapper,bwa_mapperandSTAR_mappermodules.
Workflow Schema
Requires
fastq files. Either paired end or Single end.
Programs required
Example of Sample File
Title Paired_end_project
#SampleID Type Path lane
Sample1 Forward /path/to/Sample1_F1.fastq.gz 1
Sample1 Forward /path/to/Sample1_F2.fastq.gz 2
Sample1 Reverse /path/to/Sample1_R1.fastq.gz 1
Sample1 Reverse /path/to/Sample1_R2.fastq.gz 2
Sample2 Forward /path/to/Sample2_F1.fastq.gz 1
Sample2 Reverse /path/to/Sample2_R1.fastq.gz 1
Sample2 Forward /path/to/Sample2_F2.fastq.gz 2
Sample2 Reverse /path/to/Sample2_R2.fastq.gz 2
Download
The workflow file is available here