BLAST operations to/from an assembled genome (in FASTA format)
- Author
Menachem Sklarz
- Affiliation
Bioinformatics Core Facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
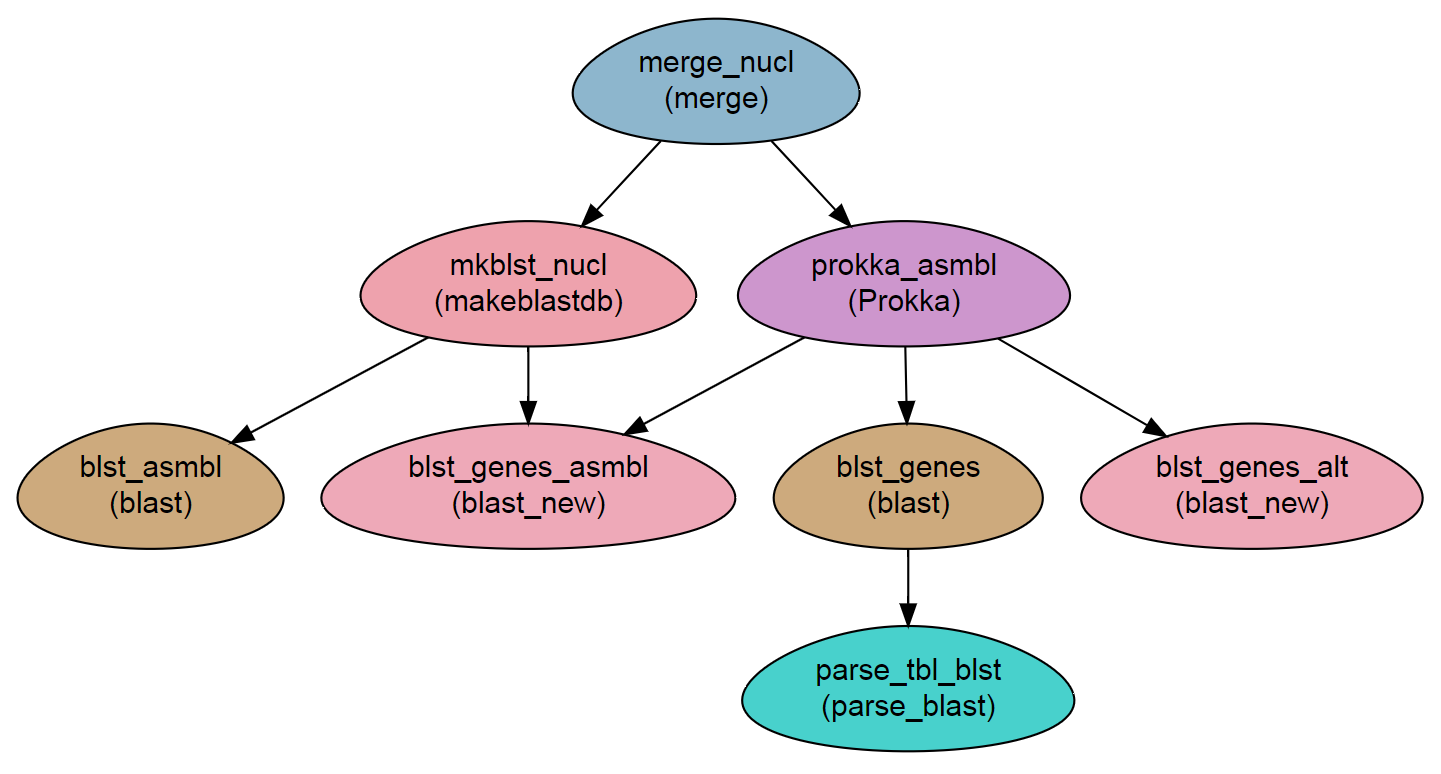
This workflow shows various ways for using BLAST.
It begins with an assembly, i.e. a file in fasta format containing genomic sequences, and performs various BLAST searches on the file.
Steps:
Merging (
merge)Constructing a BLAST db from the assembly using the
makeblastdbmoduleSearching the assembly using a fasta query file.
Annotation of the assemblies using
ProkkaUsing the resulting predicted gene sequences to search a BLAST database (
blastandparse_blastmodules).Using the alternative BLAST module (
blast_new) to search the assembly for the predicted genes.
Workflow Schema
Requires
Nucleotide fasta files.
Programs required
Example of Sample File
Title BlastExperiment
#SampleID Type Path
Sample1 Nucleotide /path/to/Sample1_.fna
Sample2 Nucleotide /path/to/Sample2_.fna
Download
The workflow file is available here