BLAST operations to/from an assembled genome (starting from raw reads)
- Author
Menachem Sklarz
- Affiliation
Bioinformatics Core Facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
This workflow demonstrates various searching schemes with blast modules.
It begins with building an assembly from the reads, and then performs various operations on the assembly: (a) construction of a BLAST database from the assembly and searching the database with a fasta file supplied by the user; (b) annotation of the assembly and searching an external database with the predicted protein sequences and (c) searching for the predicted gene sequences in the assembly database.
Steps:
Merging and QC
Assembly using MEGAHIT (
megahitmodules at sample scope. i.e. one assembly per sample)Constructing a BLAST db from the assembly using the
makeblastdbmoduleSearching the assembly using a fasta query file (
blastmodule).Annotation of the assemblies using
Prokka.Using the resulting predicted gene sequences to search a BLAST database.
Using the alternative BLAST module (
blast_new) to search the assembly for the predicted genes.
Workflow Schema
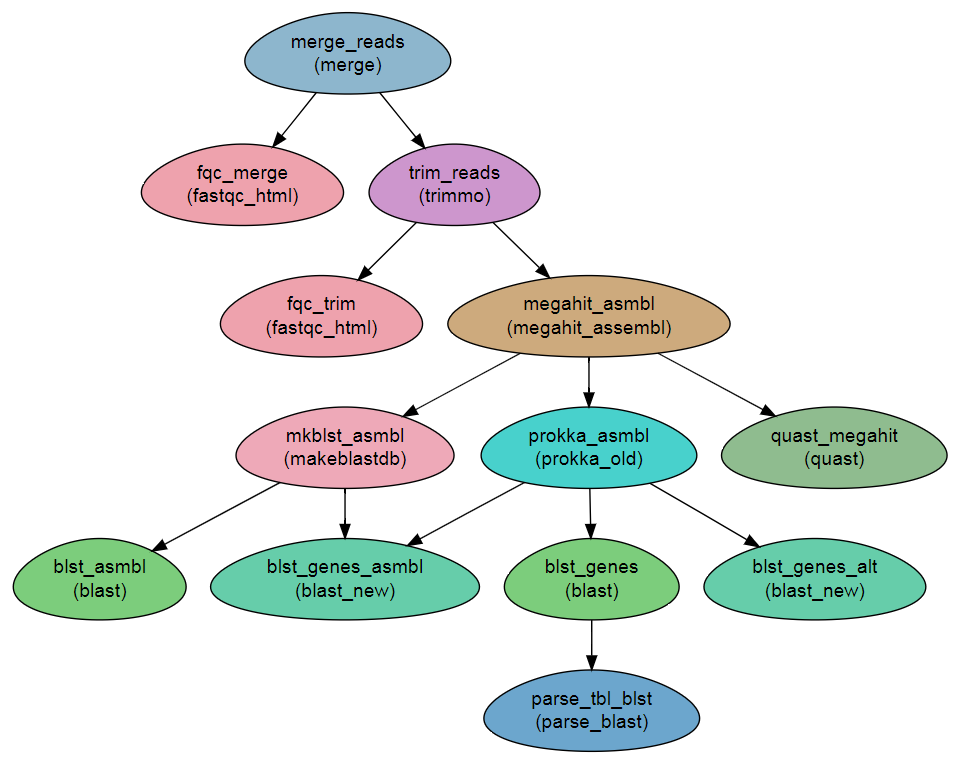
Requires
fastqc files. Either paired end or single end.
Programs required
Example of Sample File
Title Paired_end_project
#SampleID Type Path lane
Sample1 Forward /path/to/Sample1_F1.fastq.gz 1
Sample1 Forward /path/to/Sample1_F2.fastq.gz 2
Sample1 Reverse /path/to/Sample1_R1.fastq.gz 1
Sample1 Reverse /path/to/Sample1_R2.fastq.gz 2
Sample2 Forward /path/to/Sample2_F1.fastq.gz 1
Sample2 Reverse /path/to/Sample2_R1.fastq.gz 1
Sample2 Forward /path/to/Sample2_F2.fastq.gz 2
Sample2 Reverse /path/to/Sample2_R2.fastq.gz 2
Download
The workflow file is available here