Read clustering using VSEARCH
- Author
Menachem Sklarz
- Affiliation
Bioinformatics Core Facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
This workflow demonstrates methods to cluster reads with the vsearch module.
Steps:
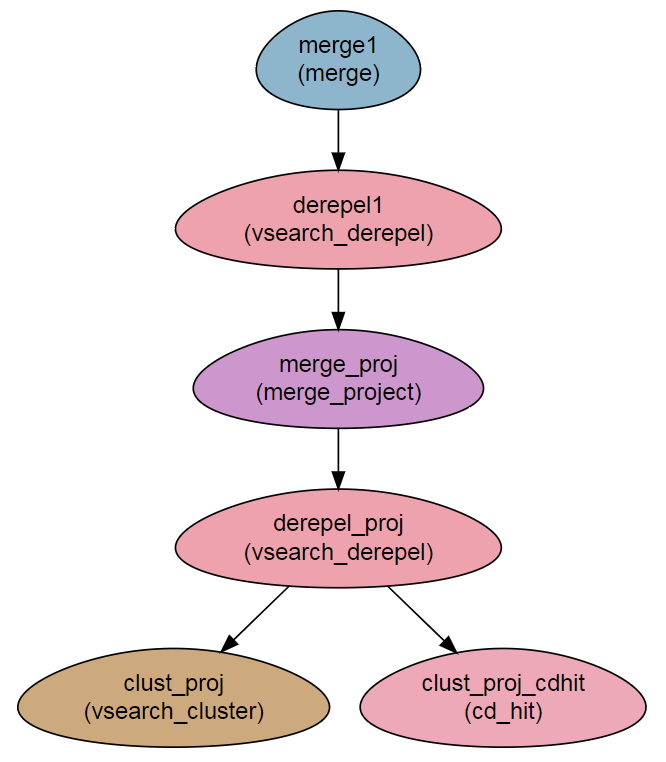
fastq files are dereplicated with
vsearchat the sample scope (vsearchproduces a `fasta` file),resulting unique sequences are merged to obtain a project-level fasta file,
project level fasta file is again dereplicated and
resulting sequences are clustered using
vsearchandcd-hit(the user can choose between them)
Workflow Schema
Requires
fastq files, either paired-end or single-end.
Programs required
Example of Sample File
Title ChIP_project
#SampleID Type Path lane
Sample1 Forward /path/to/Sample1_F1.fastq.gz 1
Sample1 Forward /path/to/Sample1_F2.fastq.gz 2
Sample1 Reverse /path/to/Sample1_R1.fastq.gz 1
Sample1 Reverse /path/to/Sample1_R2.fastq.gz 2
Sample2 Forward /path/to/Sample2_F1.fastq.gz 1
Sample2 Reverse /path/to/Sample2_R1.fastq.gz 1
Sample2 Forward /path/to/Sample2_F2.fastq.gz 2
Sample2 Reverse /path/to/Sample2_R2.fastq.gz 2
Download
The workflow file is available here