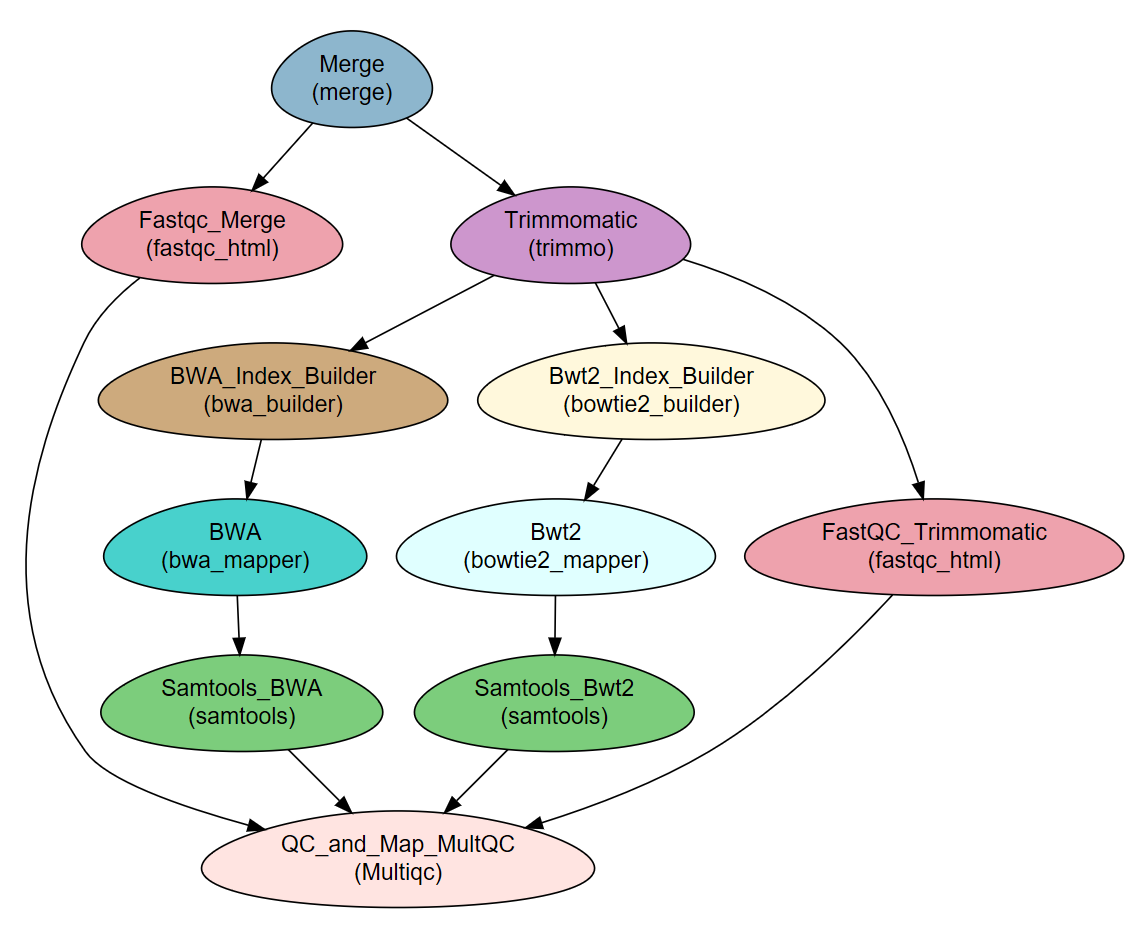
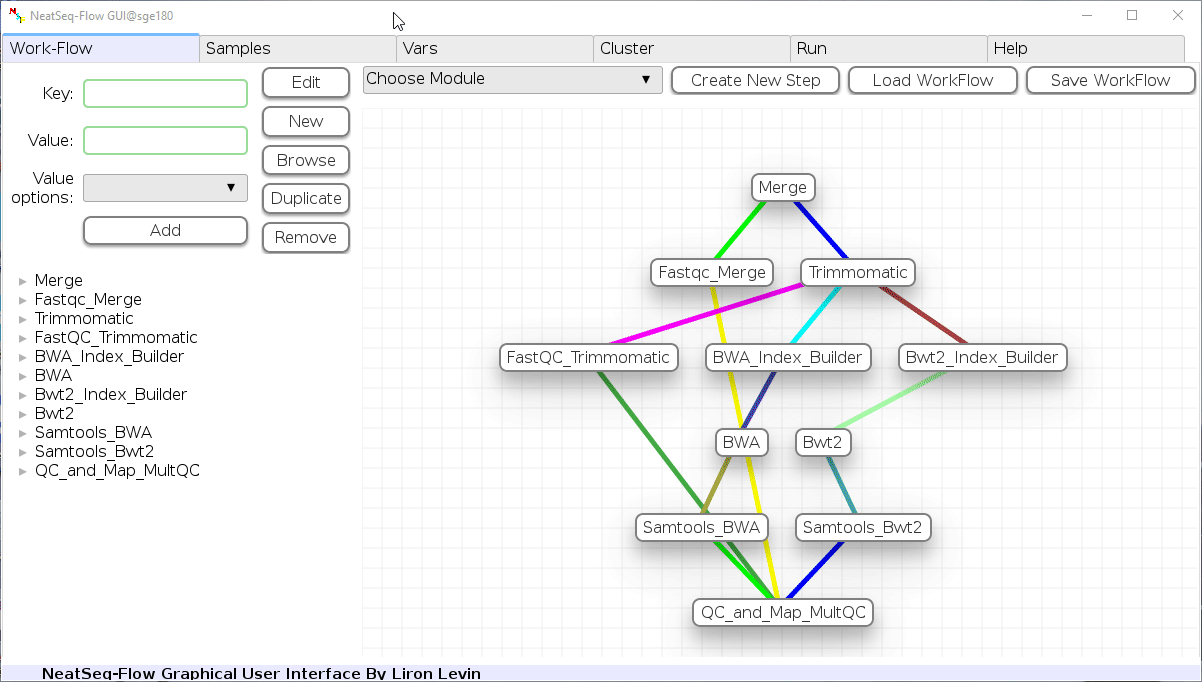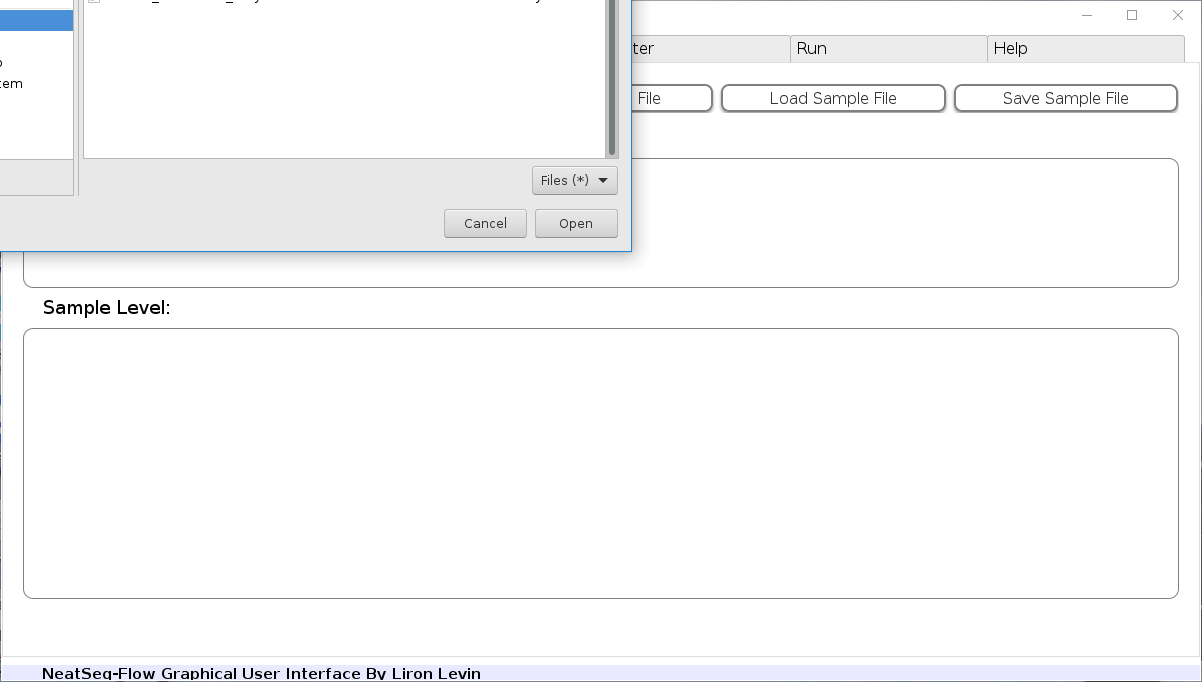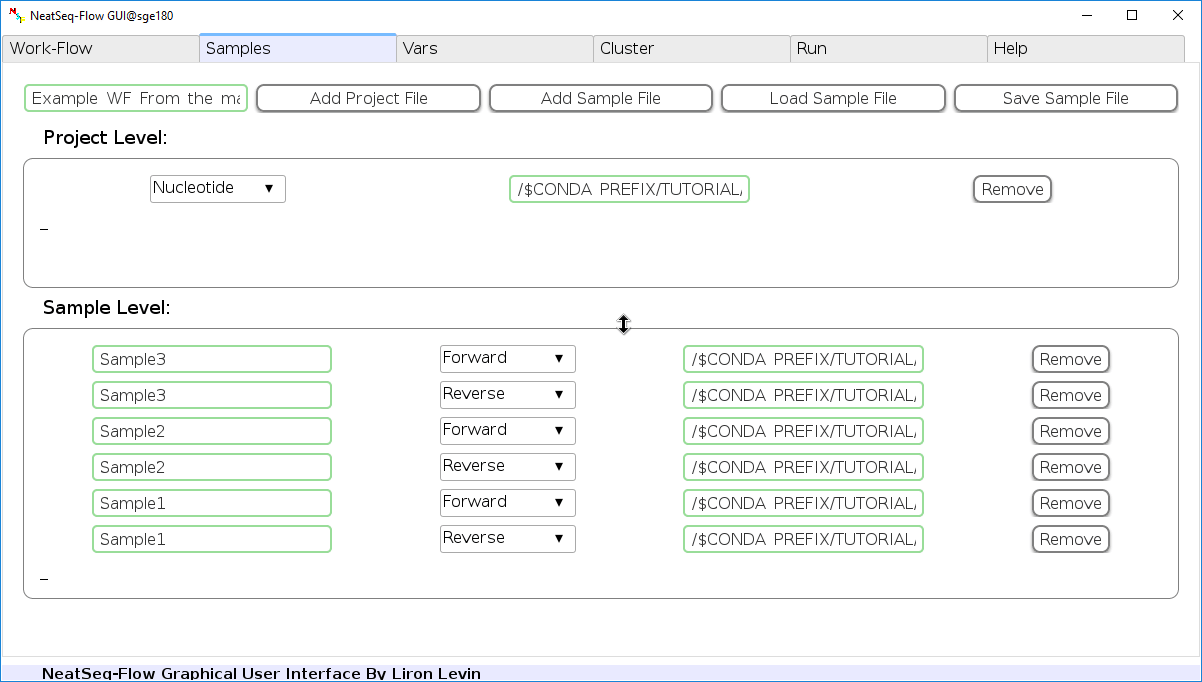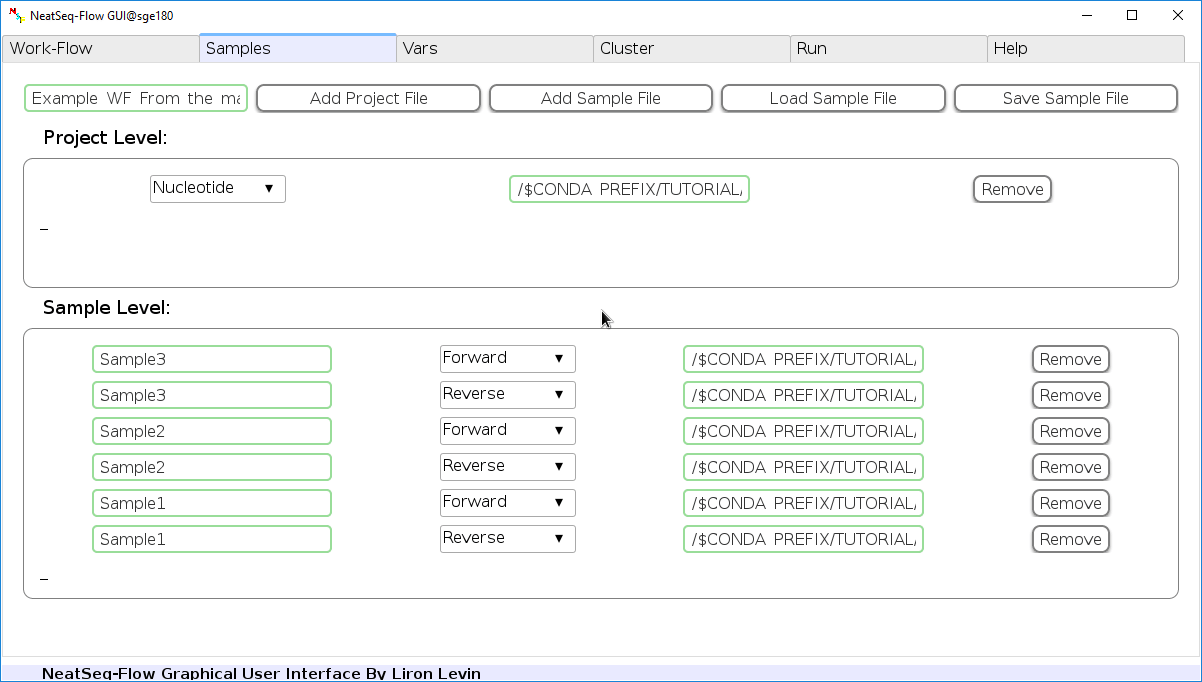
The example workflow receives FASTQ files and performs:
Quality testing and trimming of the raw sequence reads (paired- or single-end).
Alignment (“mapping”) of the reads to a reference genome using two different programs.
Sorting the samples’ BAM files as final results.
Creation of a report on reads and mapping quality.
The input files in this tutorial are whole genome sequenced bacteria, and the resulting BAM files may be used for subsequent variant calling and other analyses.
The example workflow is distributed as part of NeatSeq-Flow for quick testing.
This WF requires samples with fastq file(s) (paired or single) and a reference genome in fasta format.
Note
The files for the tutorial are included in the installation procedure below.
The three samples used in this example workflow are SRR453031, SRR453032 and SRR453033 from Staphylococcus aureus subsp. aureus Genome Sequencing project (BioProject PRJNA157545). The Staphylococcus aureusGCF_000284535.1 genome assembly was used as reference genome.
To save run-time and space, the raw sample files contain only the first 500,000 lines each.
The sample’s file indicate the files that will be used in this analysis.
These files were downloaded when the Tutorial’s conda environment was created in the Create the Tutorial conda environment step and are found within the conda environment itself
Typically, the installation of both NeatSeq-Flow and its GUI is done on a Linux operating system.
It is then possible to use the GUI from any computer through a web-browser.
Copy the “User Name” (blue line) from the terminal to the “User Name” form in the login window
Copy the “Password” (yellow line) from the terminal to the “Password” form in the login window
Click on the “Login” button.
Managing Users:
It is possible to mange users using SSH, NeatSeq-Flow will try to login by ssh to a host using the provided “User Name” and “Password”.
The ssh host can be local or remote.
Note: If using a remote host, NeatSeq-Flow needs to be installed on the remote host and the analysis will be run on the remote host by the user that logged-in
NeatSeq_Flow_GUI.py--Server--SSH_HOST127.0.0.1
For more option:
NeatSeq_Flow_GUI.py -h
usage: NeatSeq_Flow_GUI.py [-h] [--Server] [--PORT CHAR] [--HOST CHAR] [--SSL]
[--SSH_HOST CHAR] [--SSH_PORT CHAR] [--USER CHAR]
[--PASSW CHAR] [--USERSFILE CHAR]
[--UNLOCK_USER_DIR] [--WOKFLOW_DIR CHAR]
[--CONDA_BIN CHAR] [--LOG_DIR CHAR]
NeatSeq-Flow GUI By Liron Levin
optional arguments:
-h, --help show this help message and exit
--Server Run as Server
--PORT CHAR Use this port in which to run the app, If not set will
search for open port (Works only When --Server is set)
--HOST CHAR The host name/ip to serve the app, If not set, will try
to identify automatically (Works only When --Server is
set)
--SSL Use SSL (Only When --Server is set)
--SSH_HOST CHAR Connect using SSH to a remote host, NeatSeq-Flow needs
to be installed on the remote host (Works only When
--Server is set)
--SSH_PORT CHAR When --SSH_HOST is set use this ssh port to connect to a
remote host.
--USER CHAR User Name For This Serve (Works only When --Server is
set)
--PASSW CHAR Password For This Serve (Works only When --Server is
set)
--USERSFILE CHAR The location of a Users file in which a list of users,
E-mails addresses and Users directories are separated by
one space (as:USER user@example.com /USER/DIR). The
login password will be send to the user e-mail after
filling its user name and the password generated at the
beginning of the run (Works only When --Server is set).
You will need a Gmail account to send the password to
the users (you will be prompt to type in your Gmail
address and password)
--UNLOCK_USER_DIR Don't Lock Users to their Directory Or to the Current
Working Directory
--WOKFLOW_DIR CHAR A Path to a Directory containing work-flow files to
choose from at log-in. Works only When --Server is set.
If --SSH_HOST is set, the Path needs to be in the remote
host.
--CONDA_BIN CHAR A path to a the CONDA bin location. If --SSH_HOST is
set, the Path needs to be in the remote host.
--LOG_DIR CHAR A path to a directory to save log files about users
statistics. Only woks If --Server is set. In any way the
path needs to be at the local host.
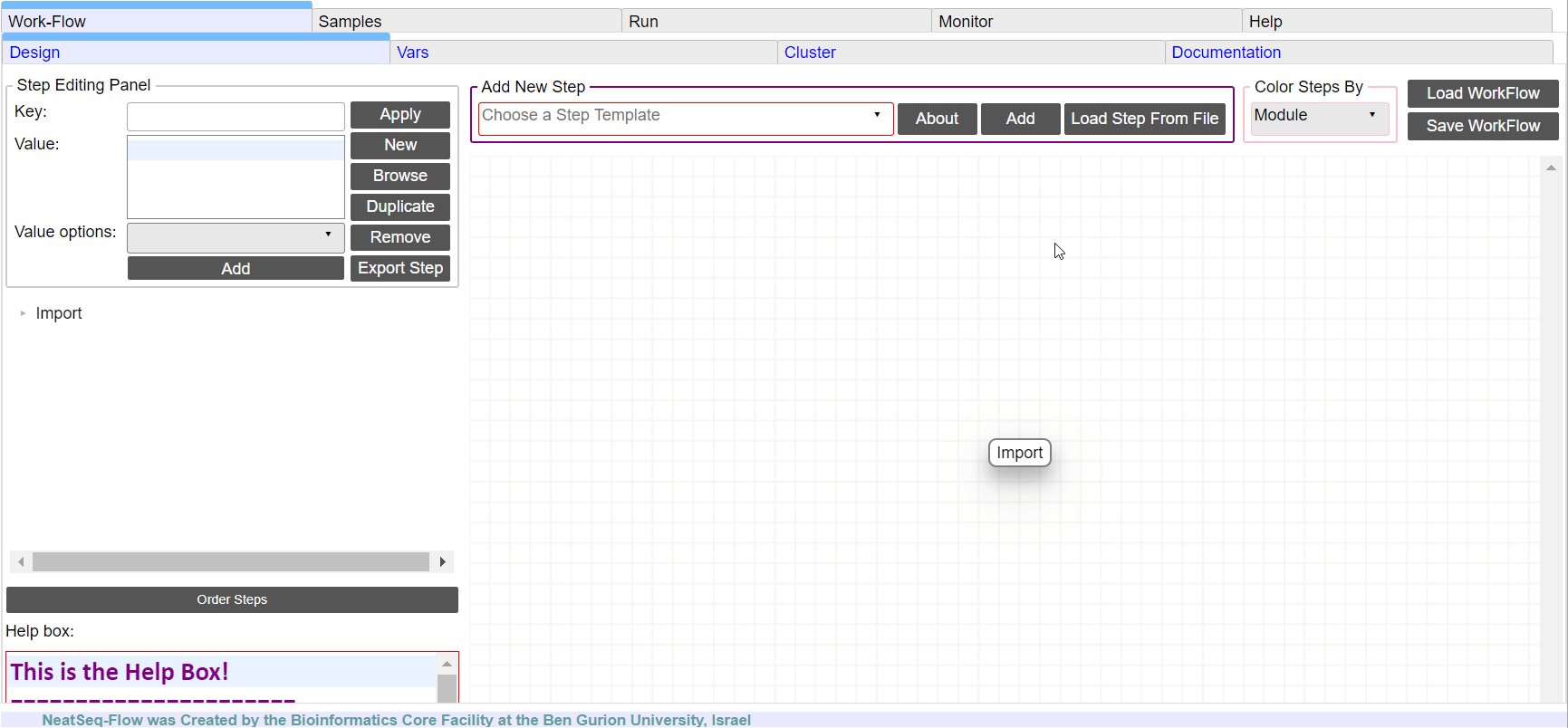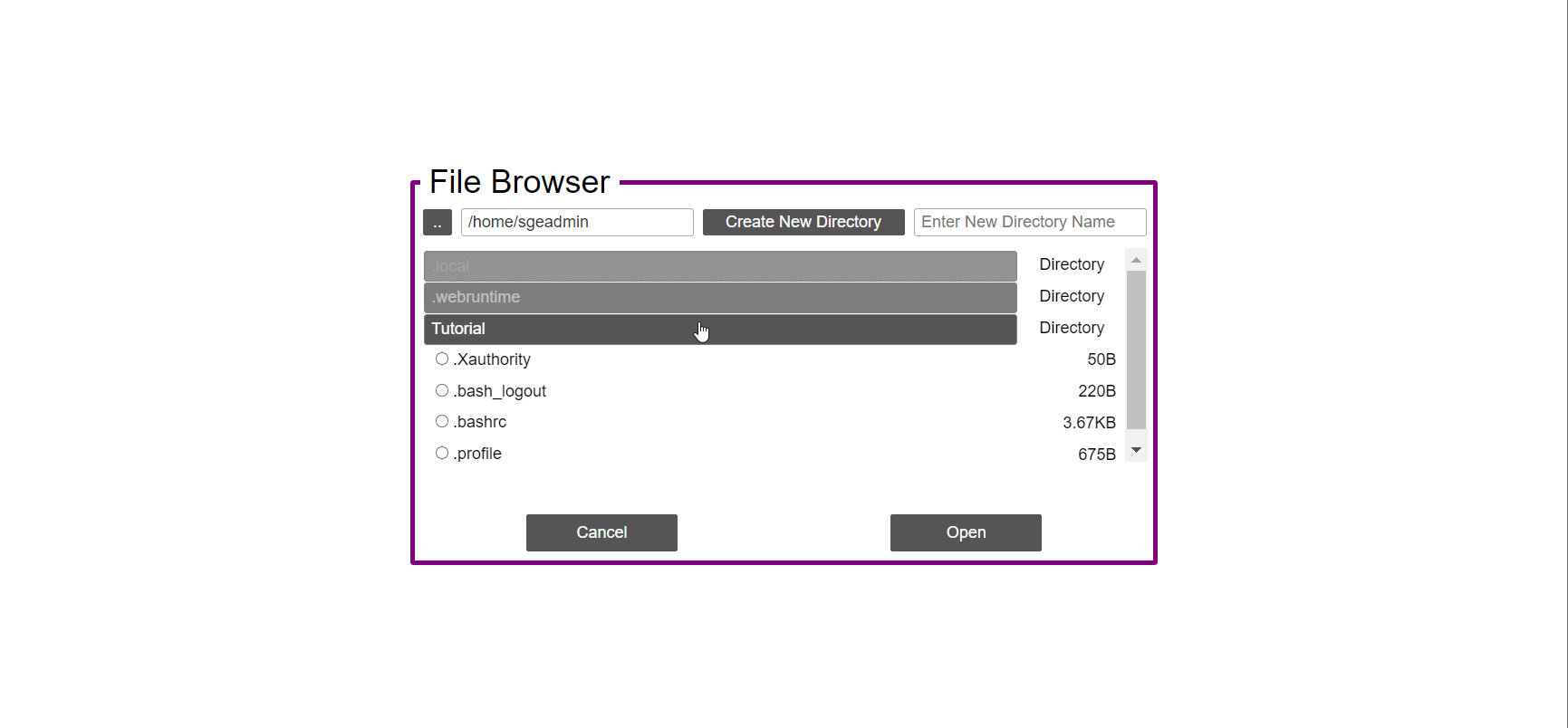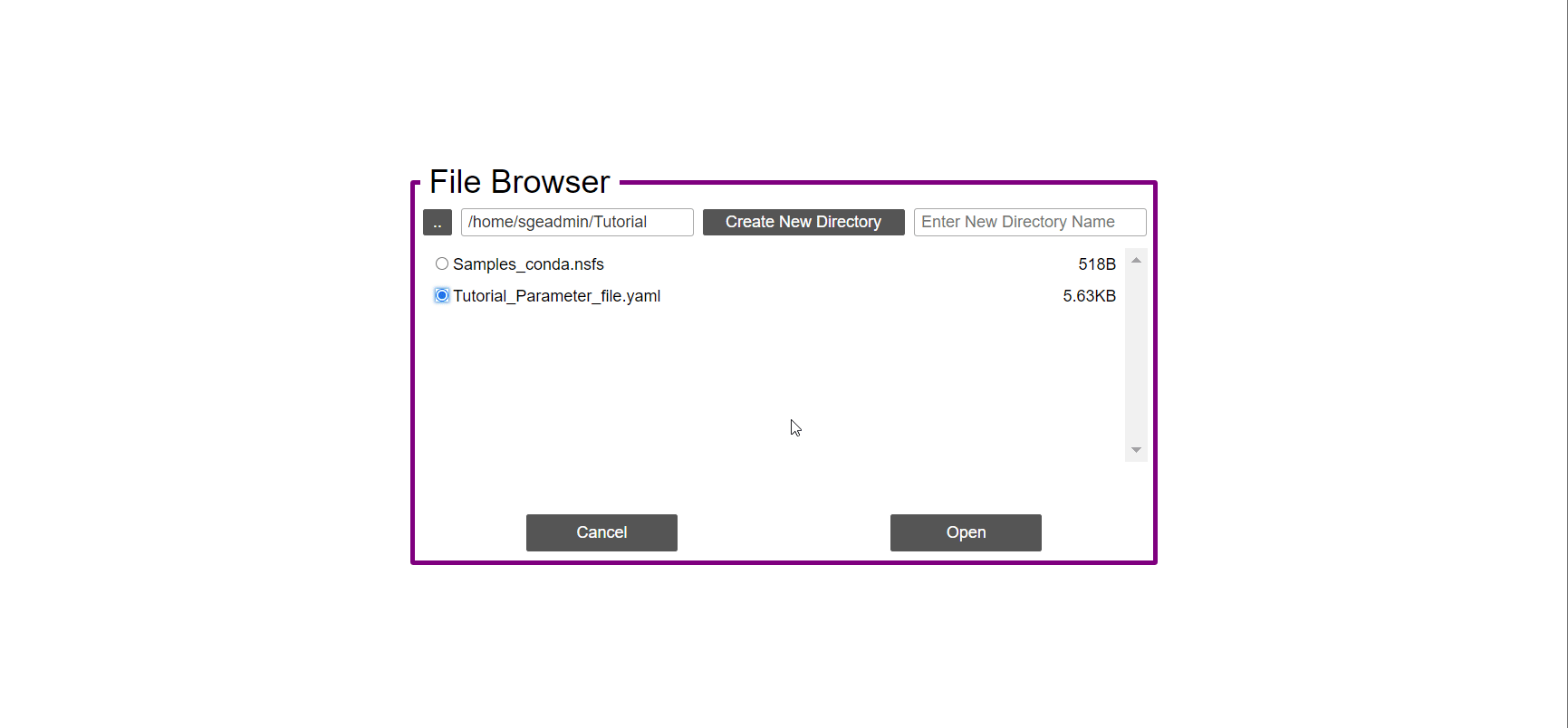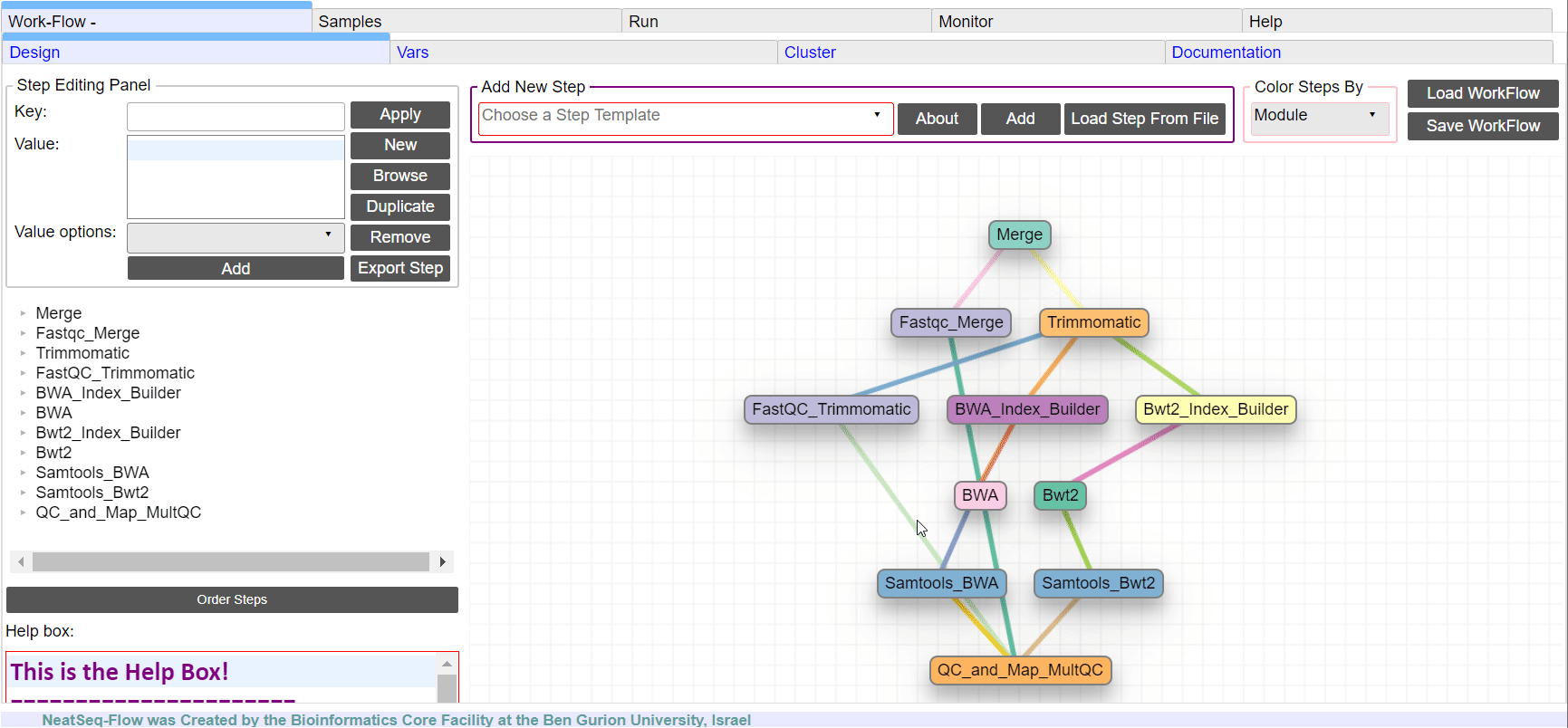
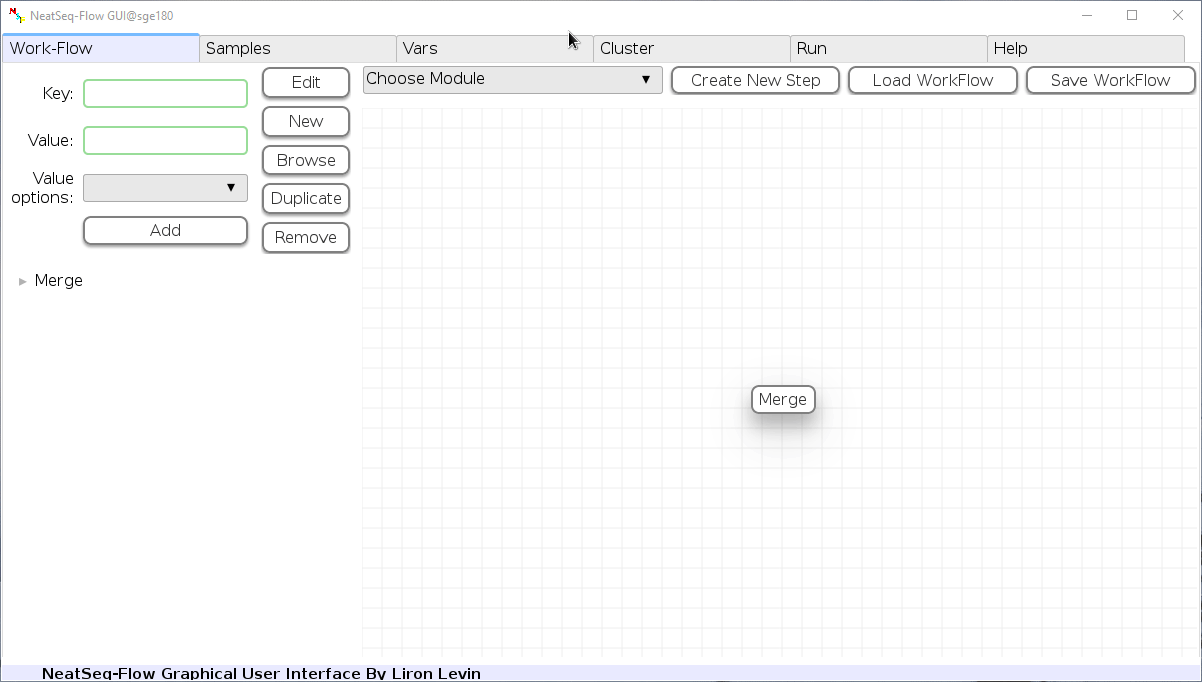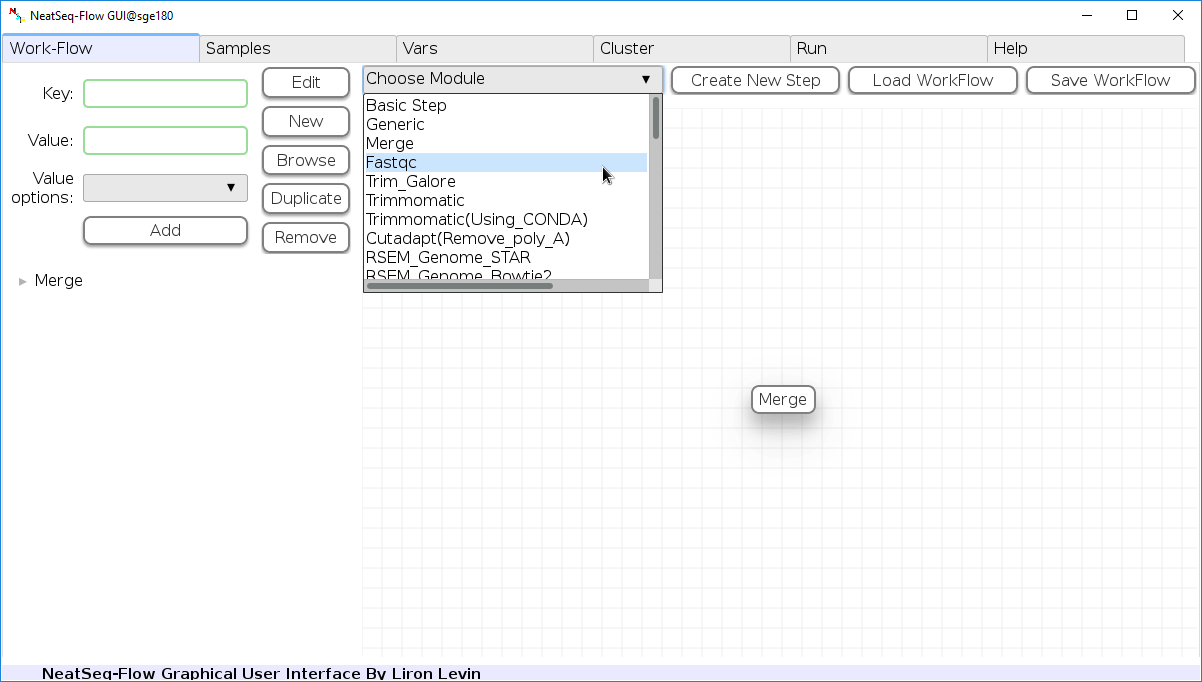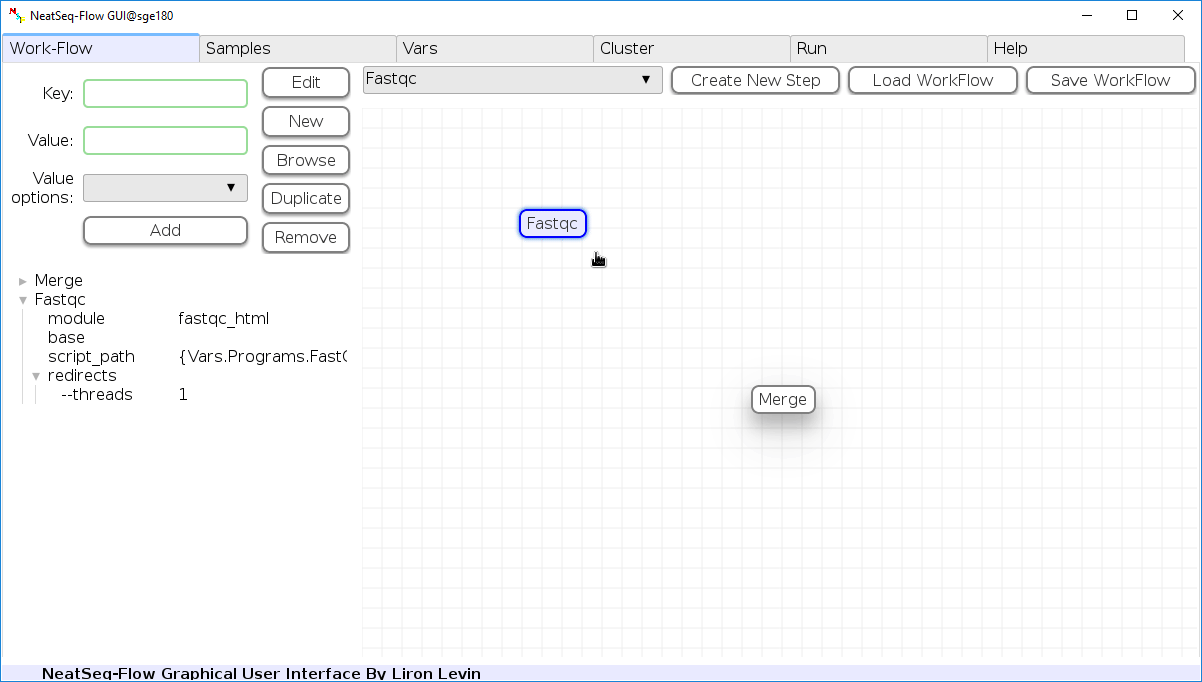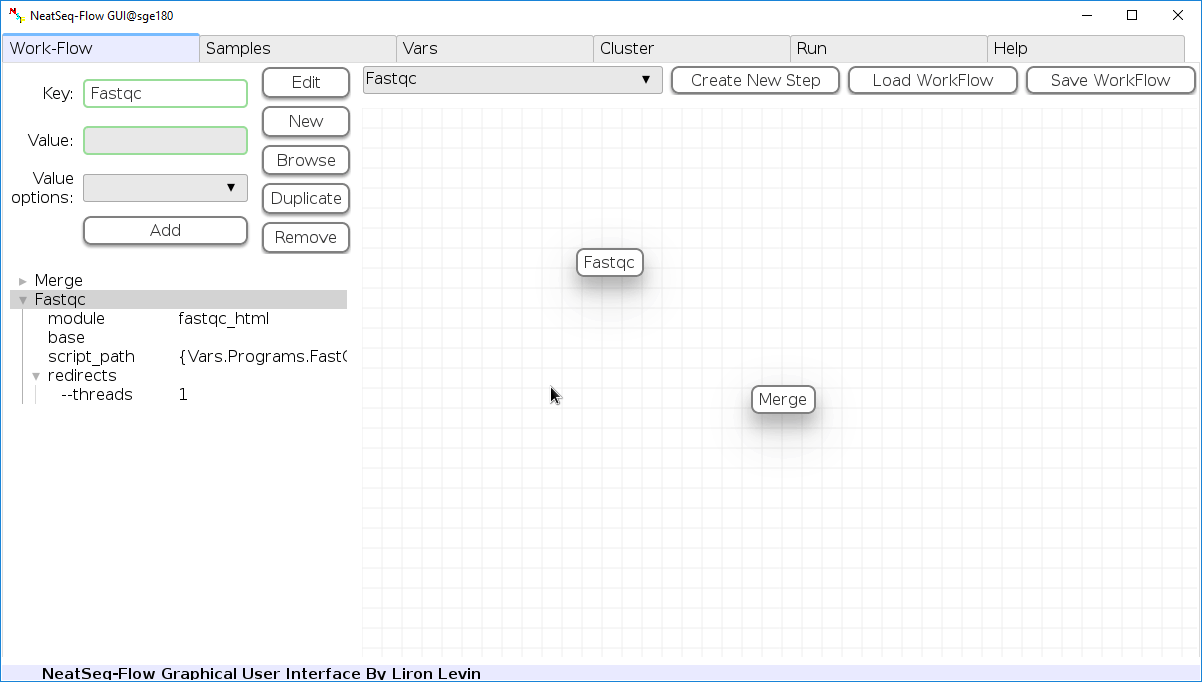
In the ‘Work-Flow’ Tab choose a module template and click on the ‘Create New Step’ button.
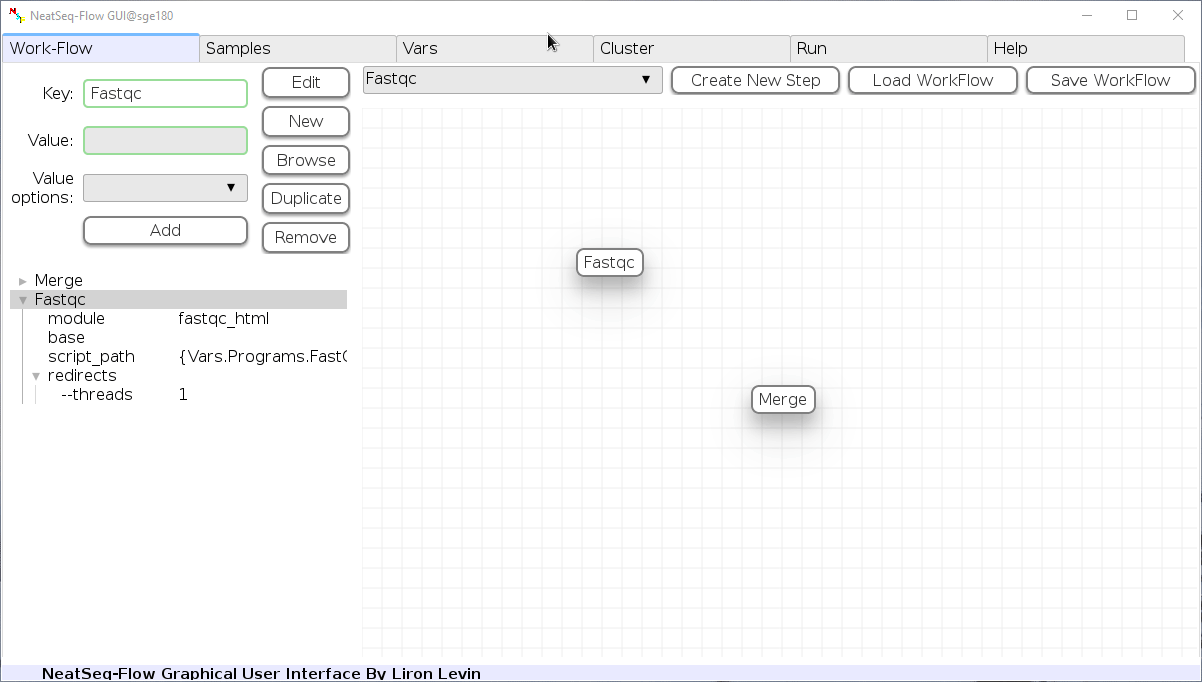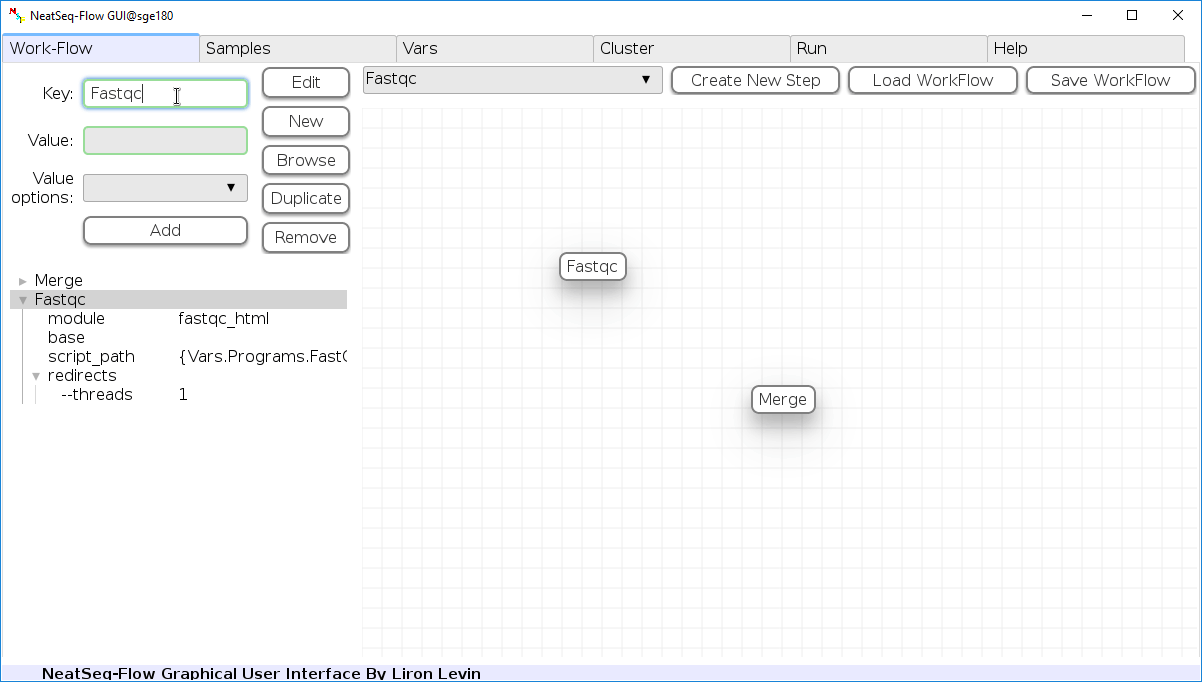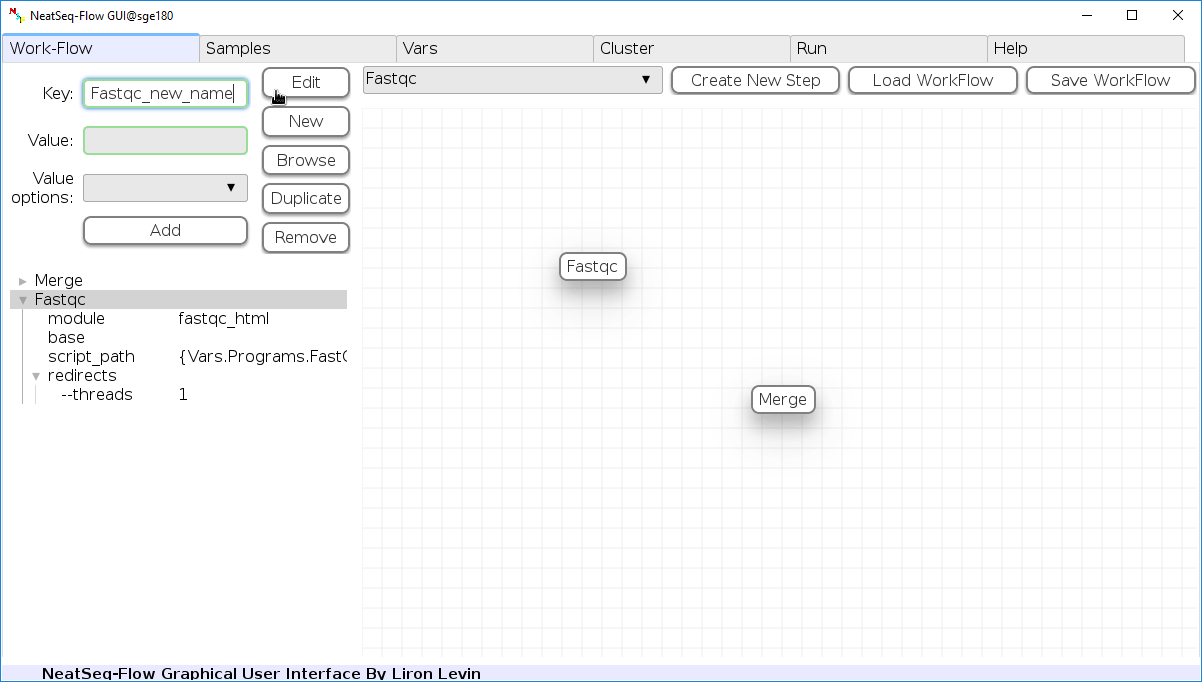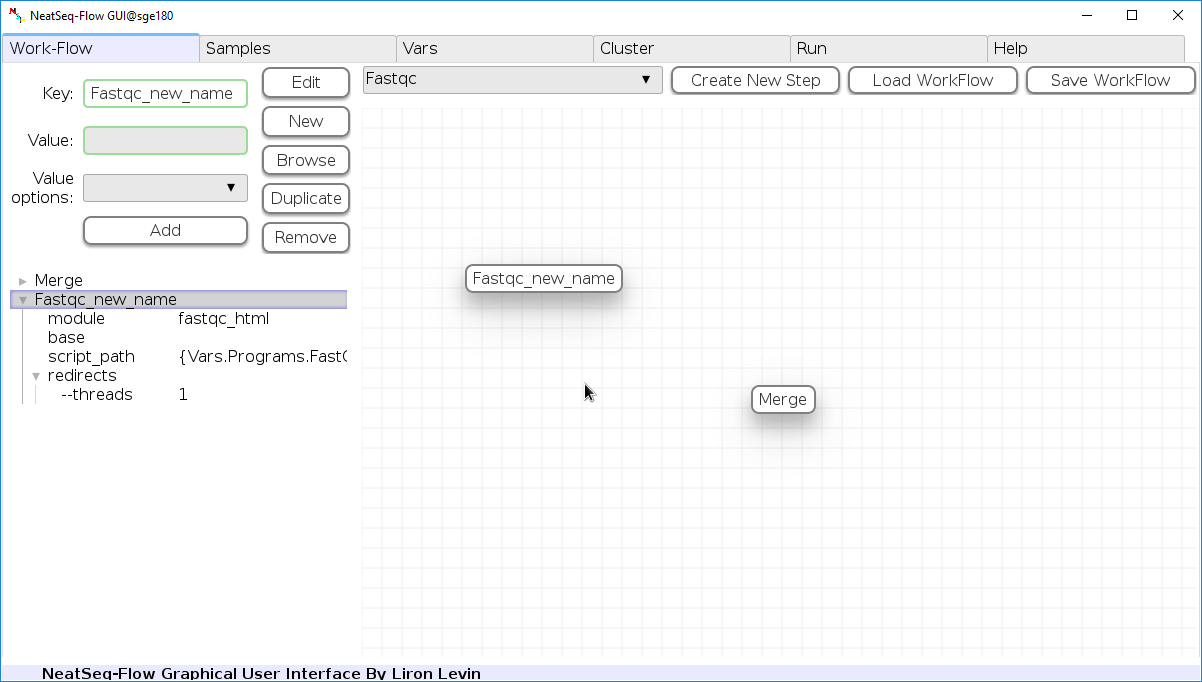
Change Step Name:
You can change the new step name by clicking on the step name and edit the key field and then click the ‘Edit’ button to set the change.
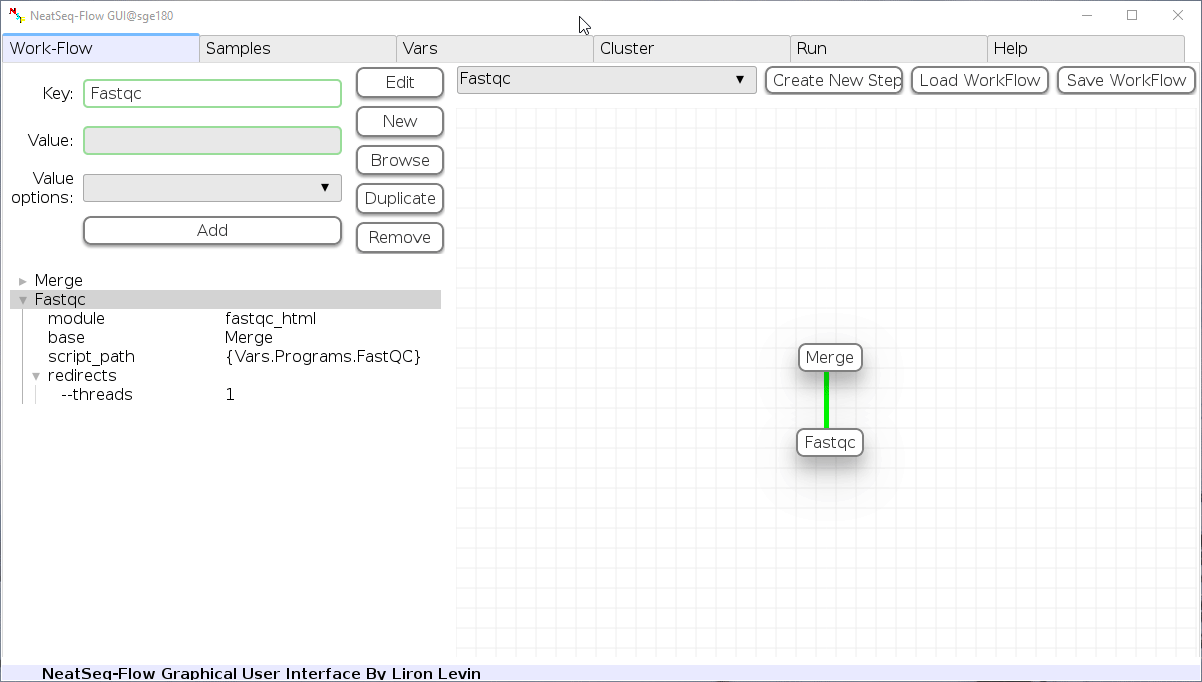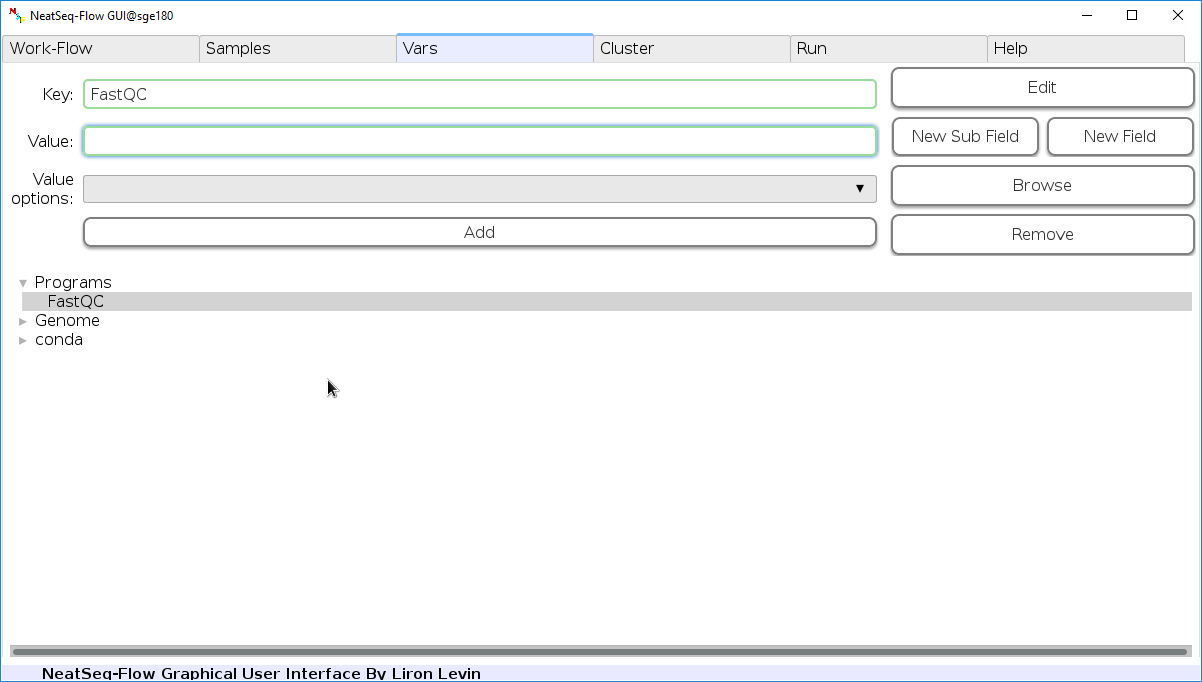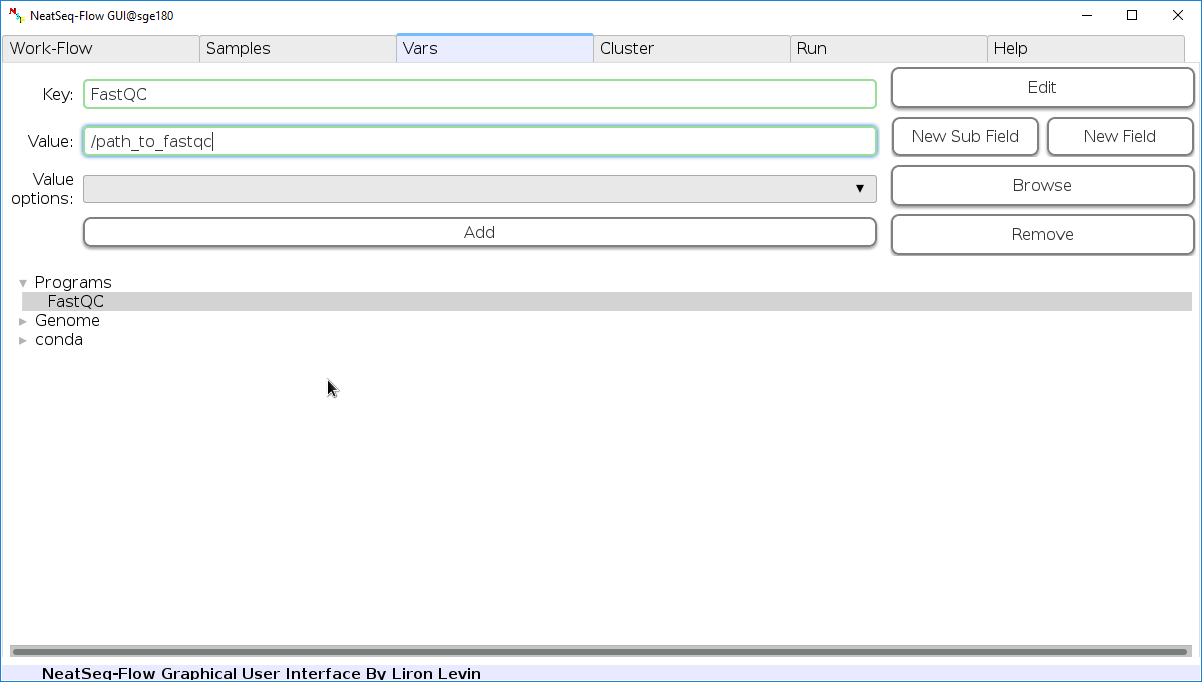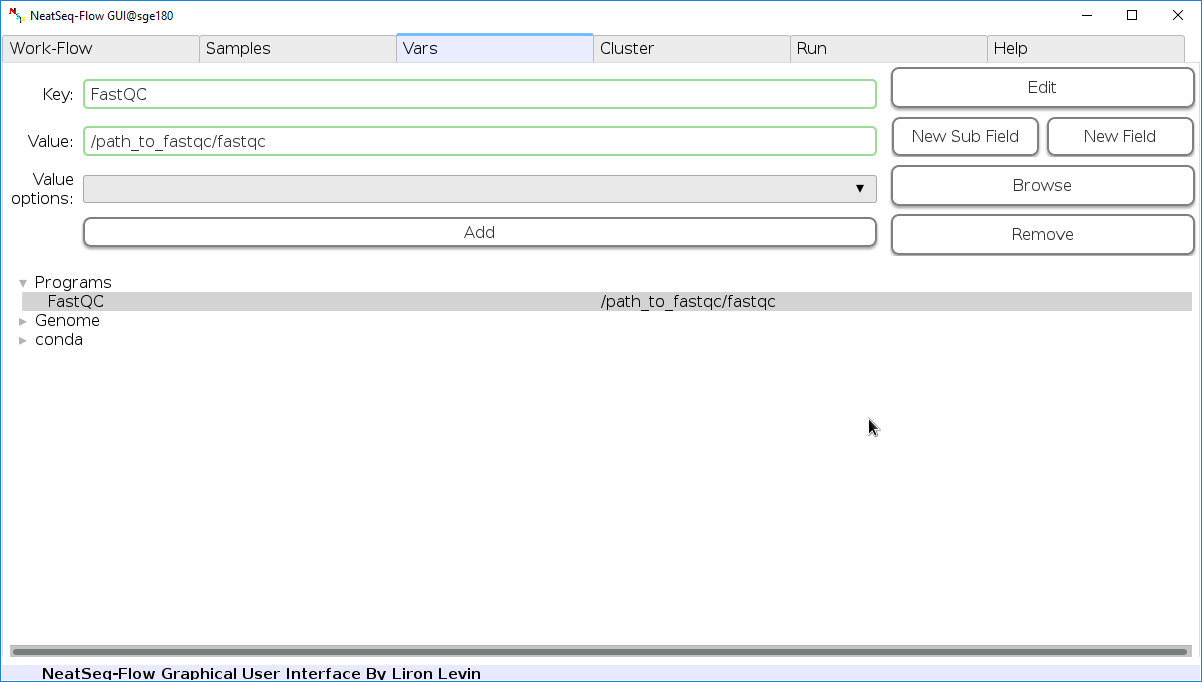
To determine the position of the new step in the work-flow:
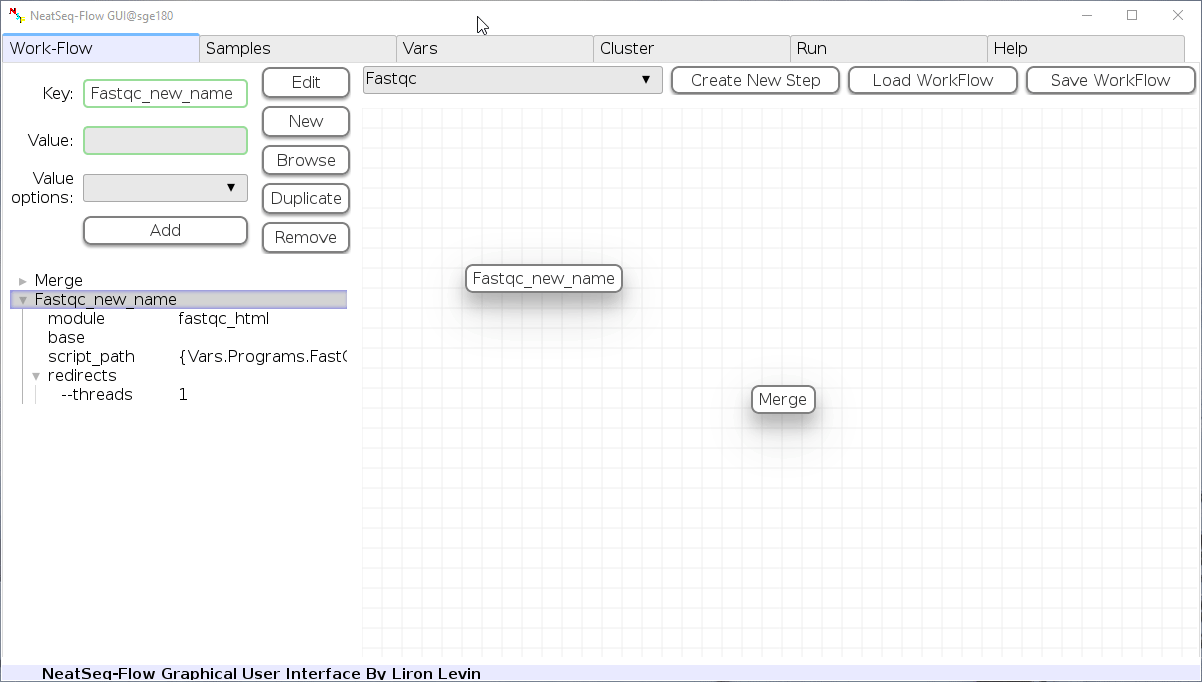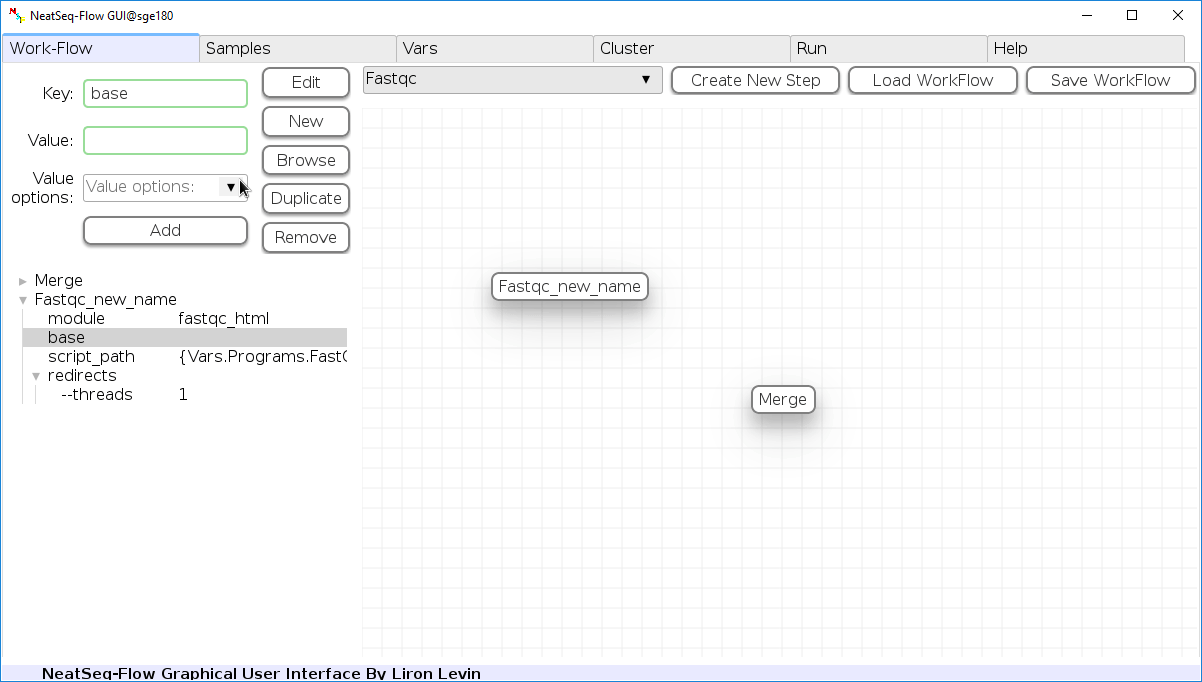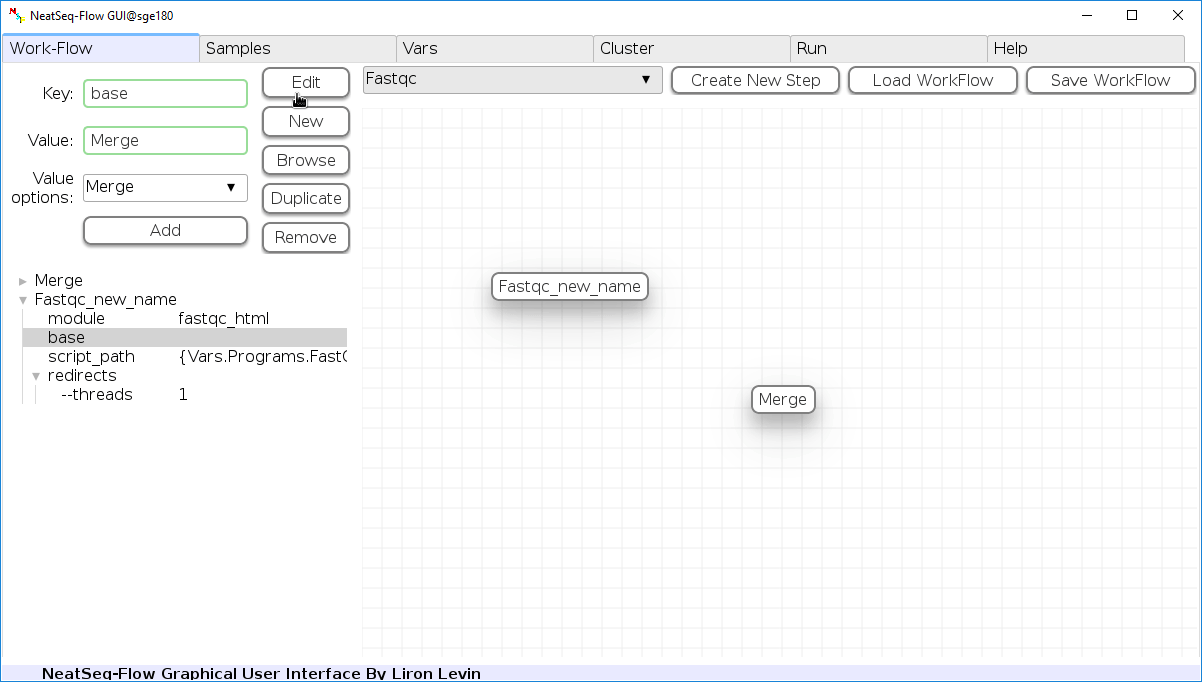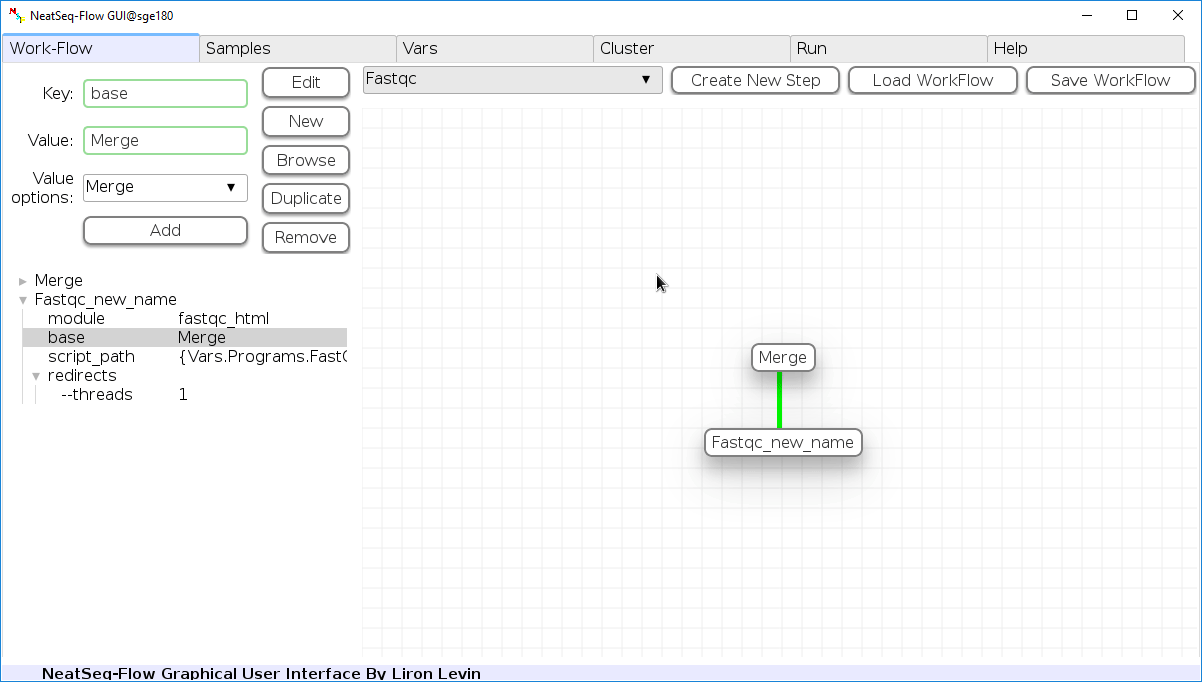
Click on the step button to see the step options
Click on the base option
Click on the ‘Value options’ drop-down menu
Choose a previous step and click the ‘Add’ button. This can be repeated to choose several previous steps.
Click the ‘Edit’ button to set the changes.
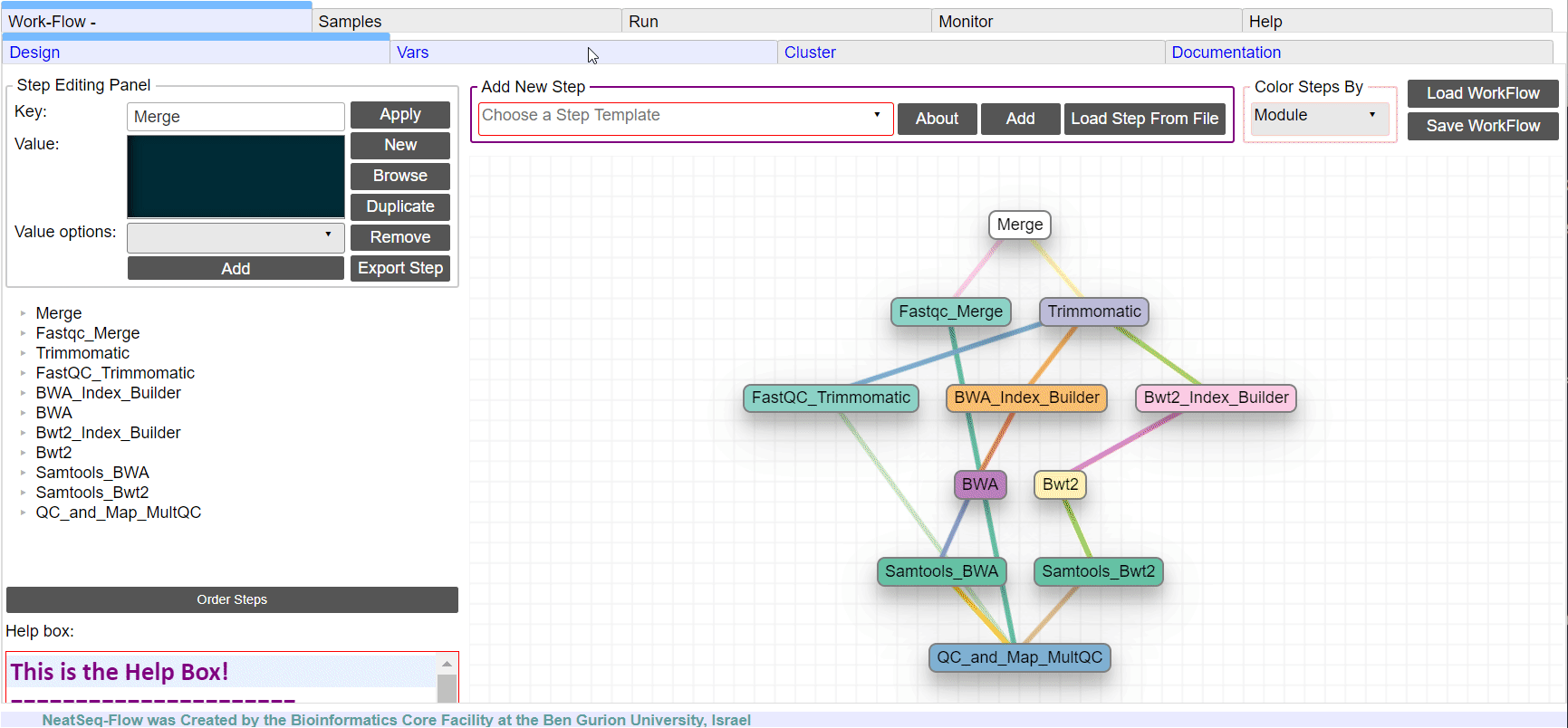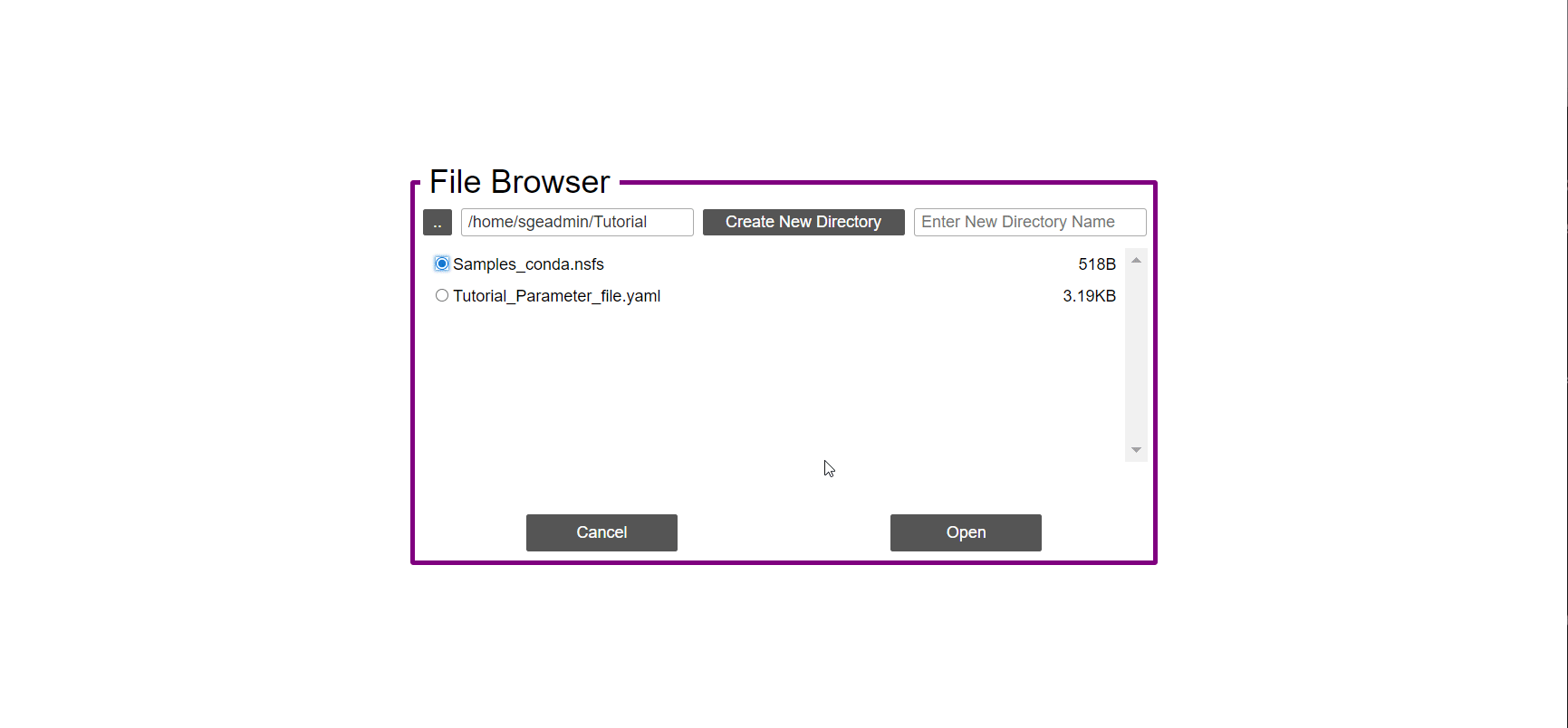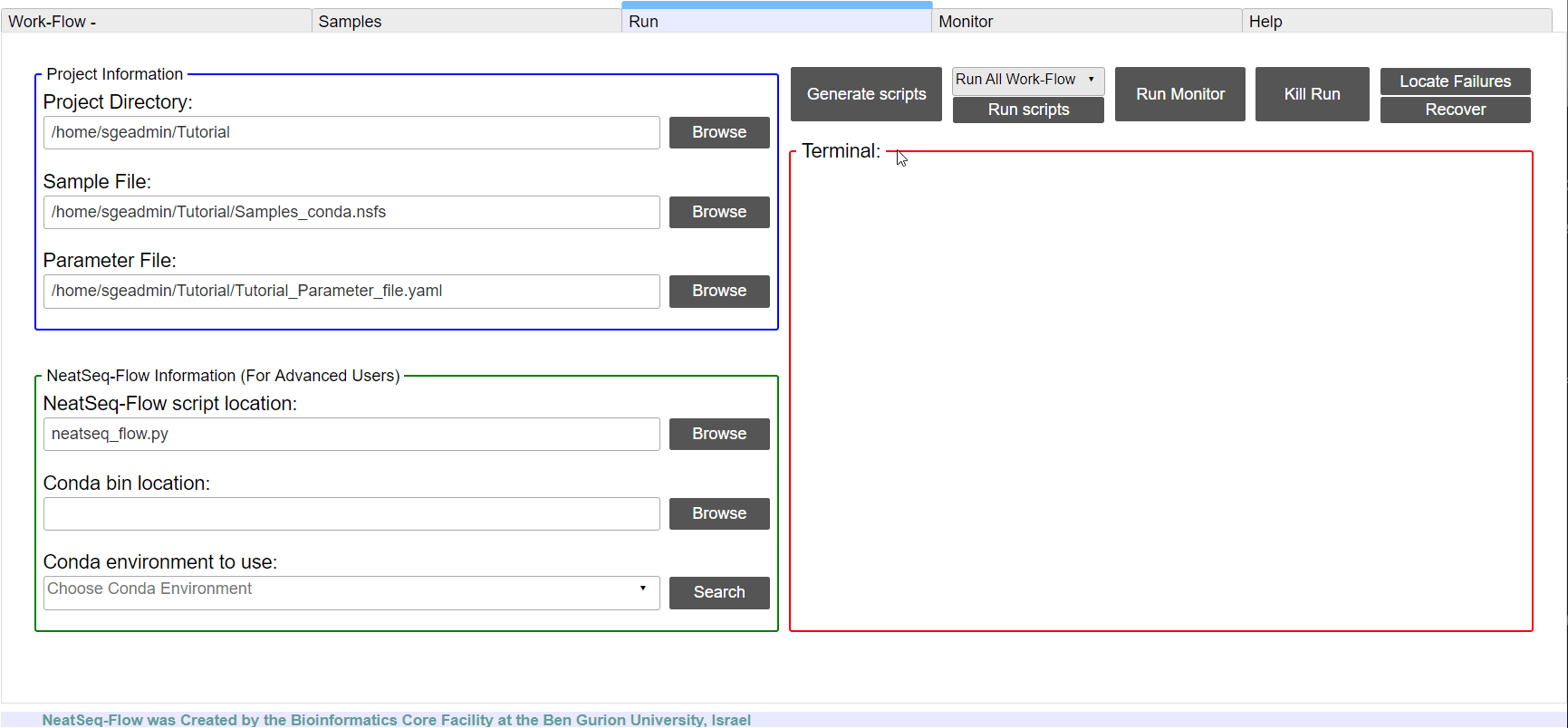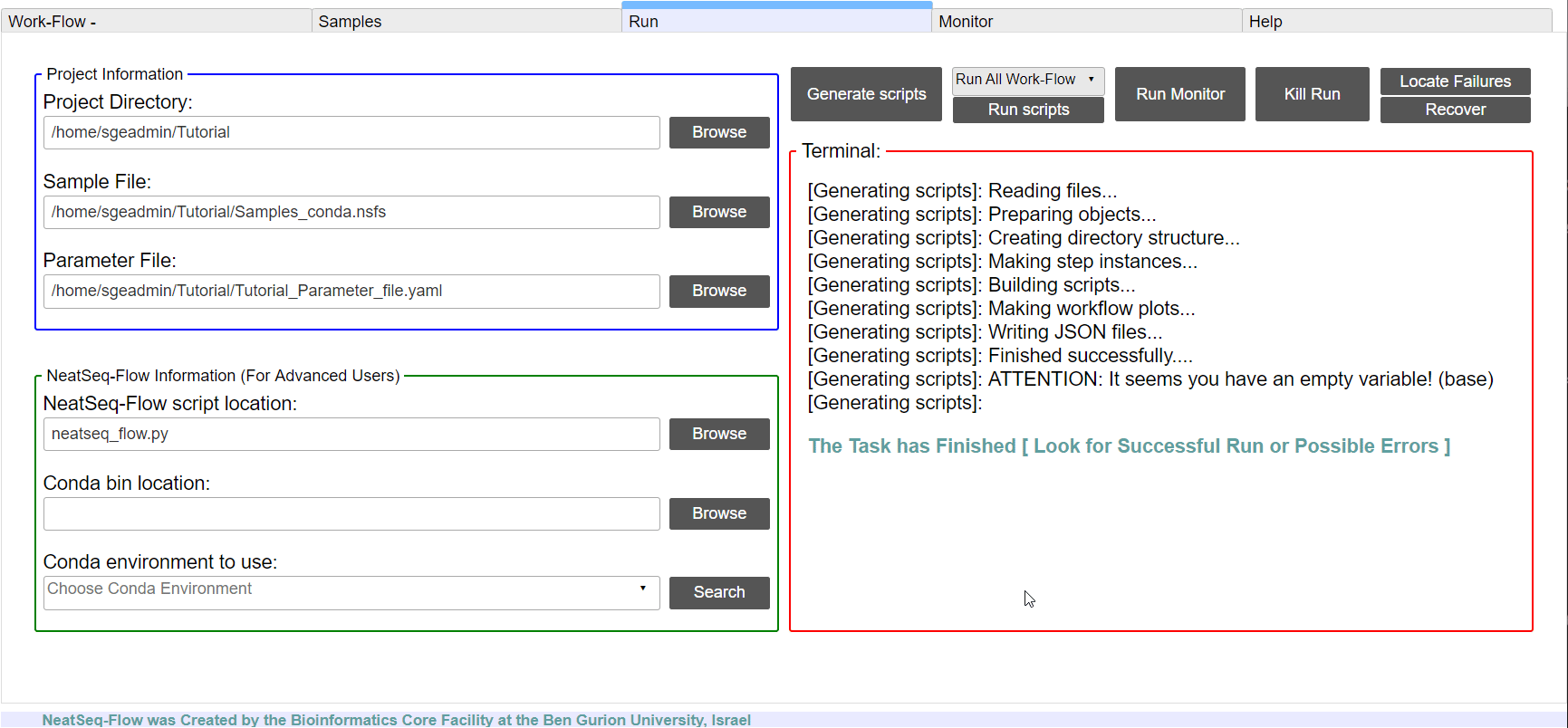
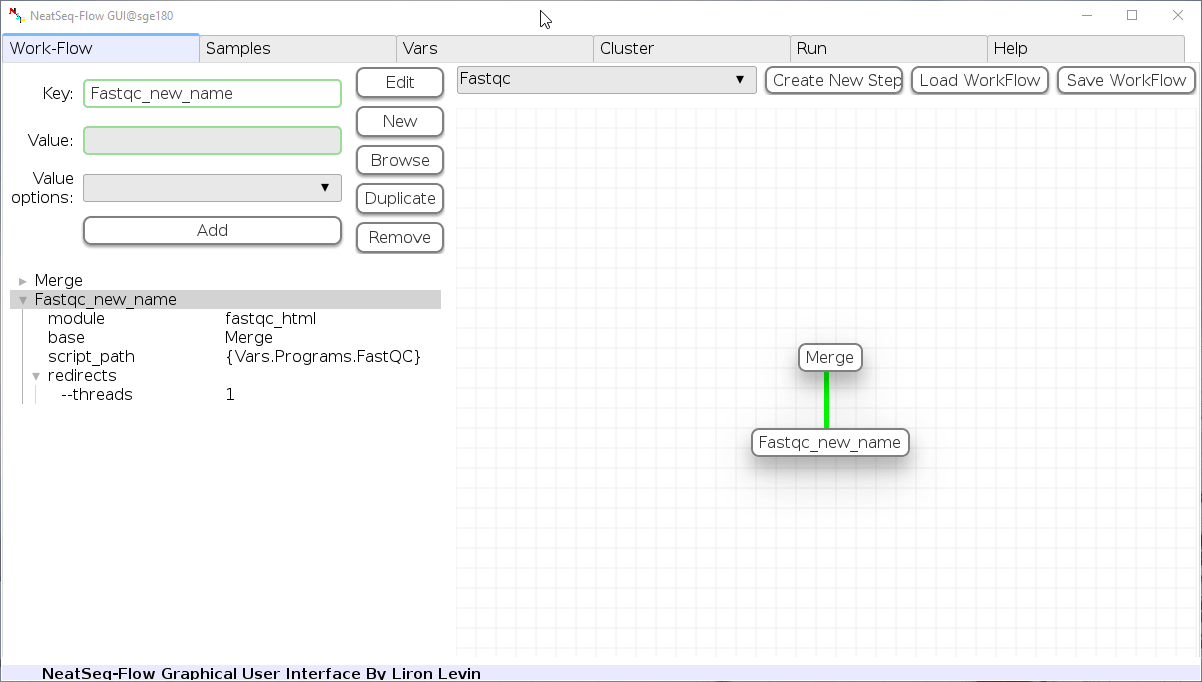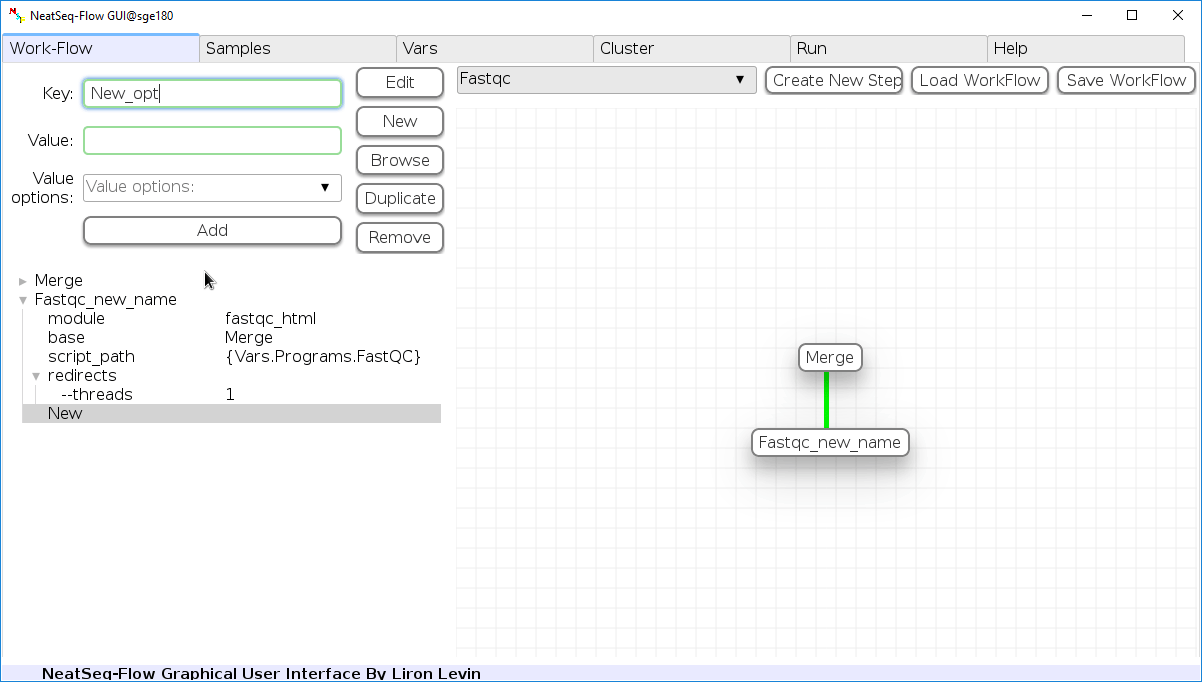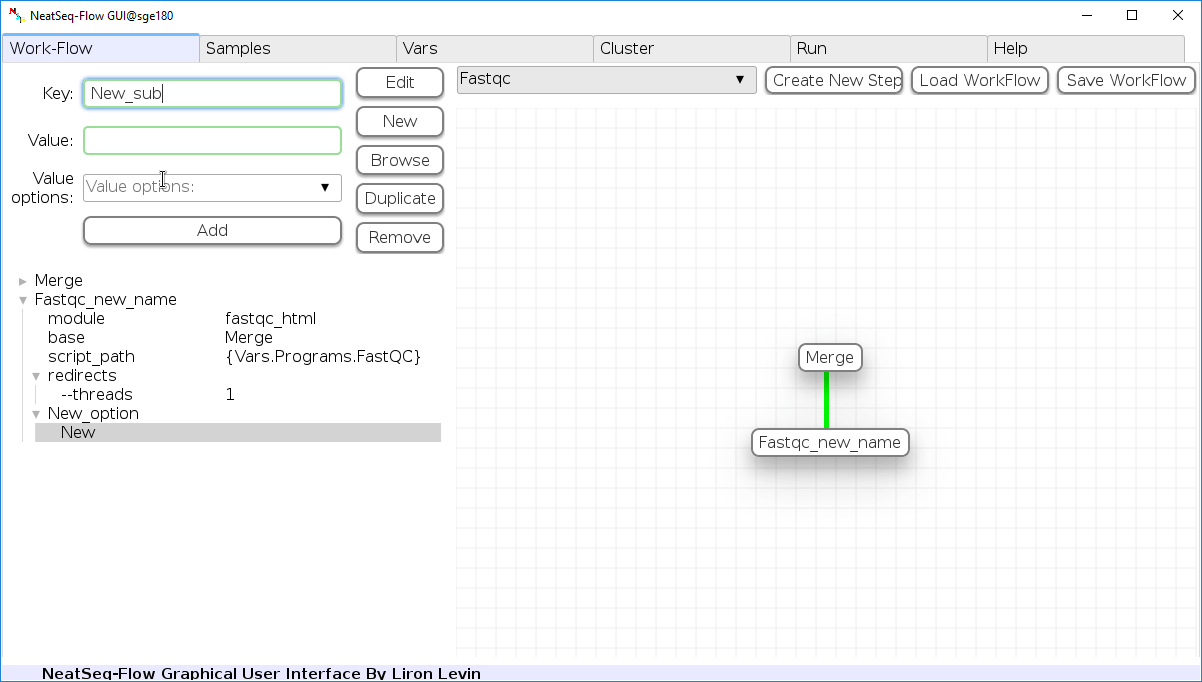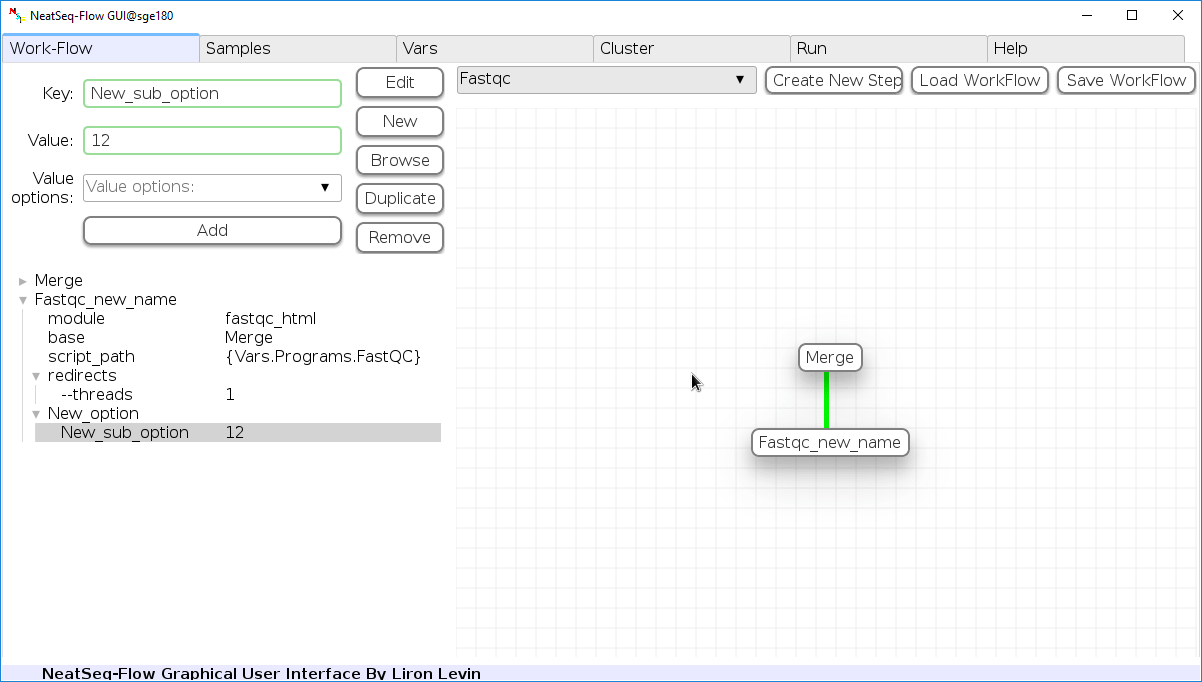
Add new step option:
Click on the step’s name (or a step option to create a new sub option)
Click on the ‘New’ button.
It is possible to edit the new option name and value by editing the ‘Key’ field and the ‘Value’ field, it is also possible to choose from the ‘Value options’ drop-down menu.
Click the ‘Edit’ button to set the changes.
Edit step’s options:
Click on the step’s option name and change the ‘Key’ field and/or the ‘Value’ field, it is also possible to choose from the ‘Value options’ drop-down menu.
When using the ‘Value options’ drop-down menu, in some cases it is possible to choose variables that are defined in the ‘Vars’ Tab.
They will appear in the form of {Vars.some_field.some_sub_field} to indicate the value found in the ‘Vars’ Tab in the some_sub_field field ( which is a sub field of ‘some_field’ ).
It is possible to choose file location as a value to the ‘Value’ field by clicking on the ‘Browse’ button.
Click the ‘Edit’ button to set the changes.
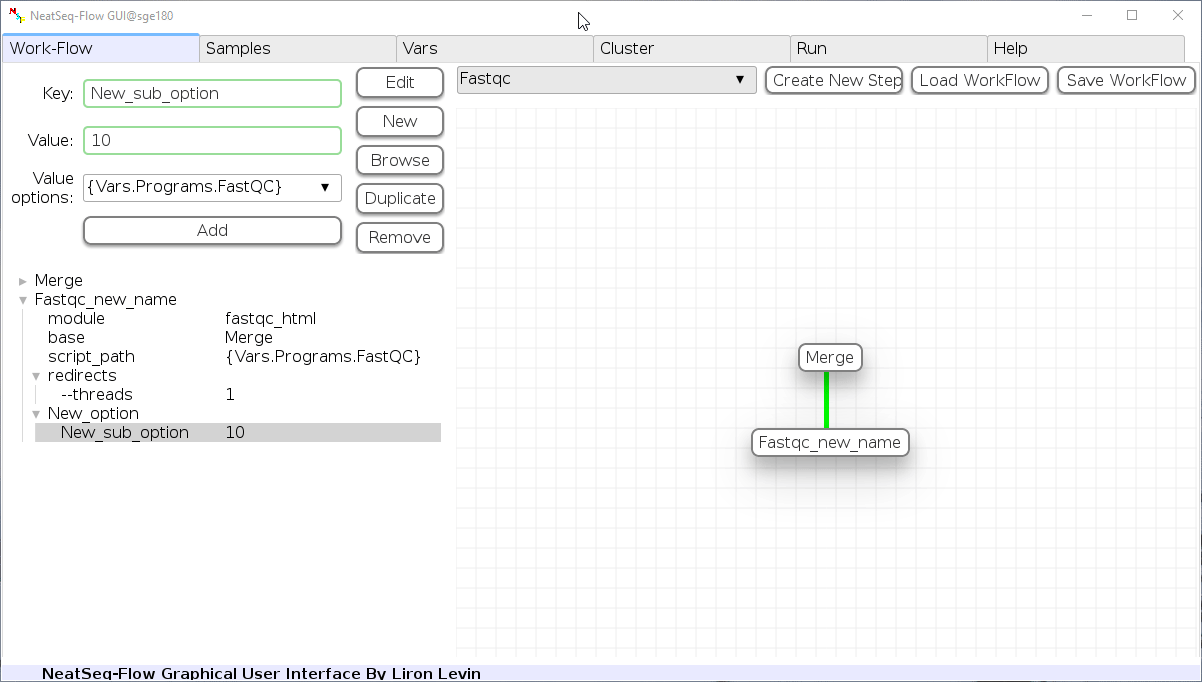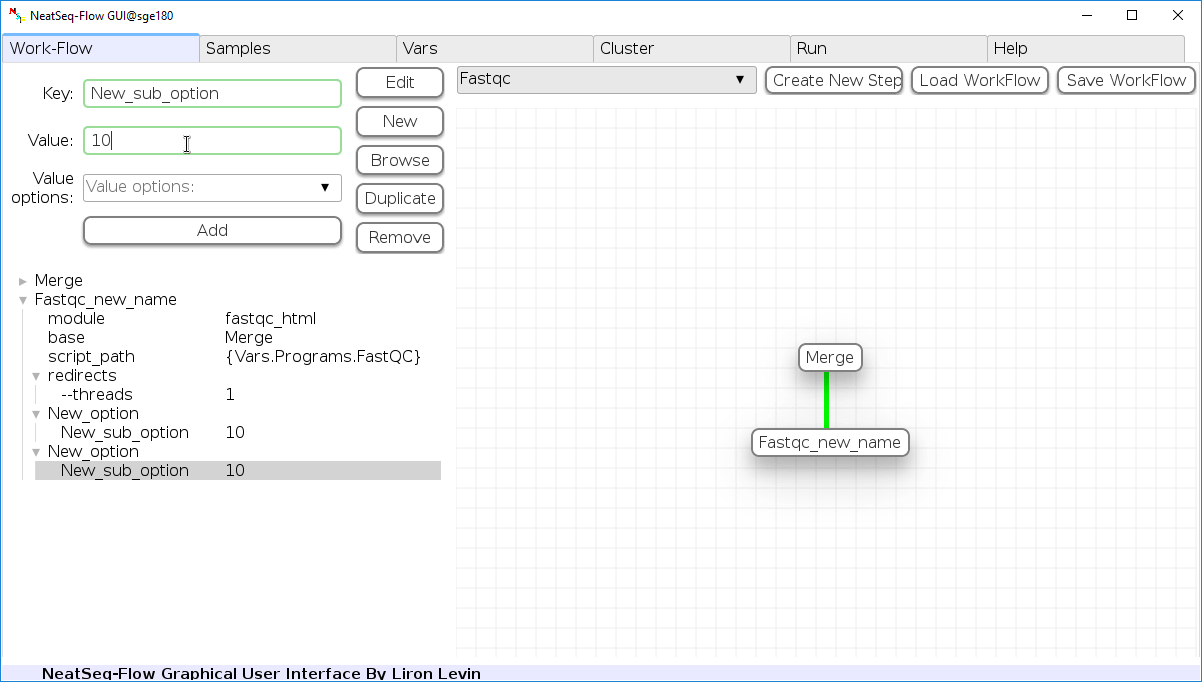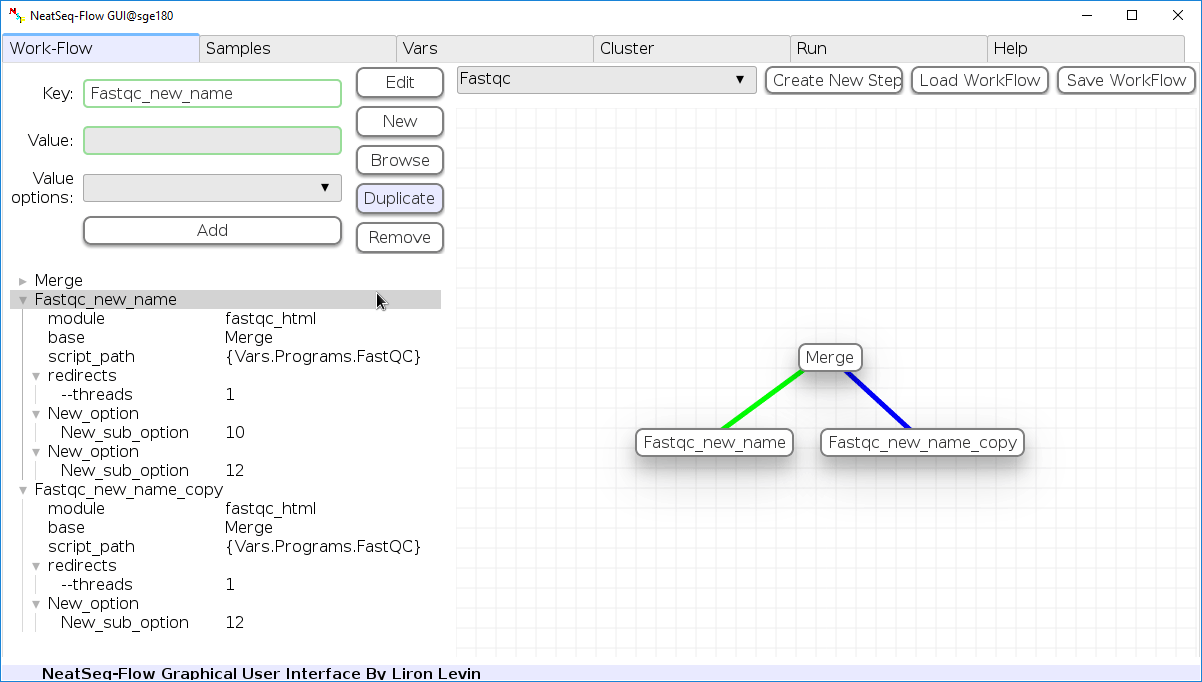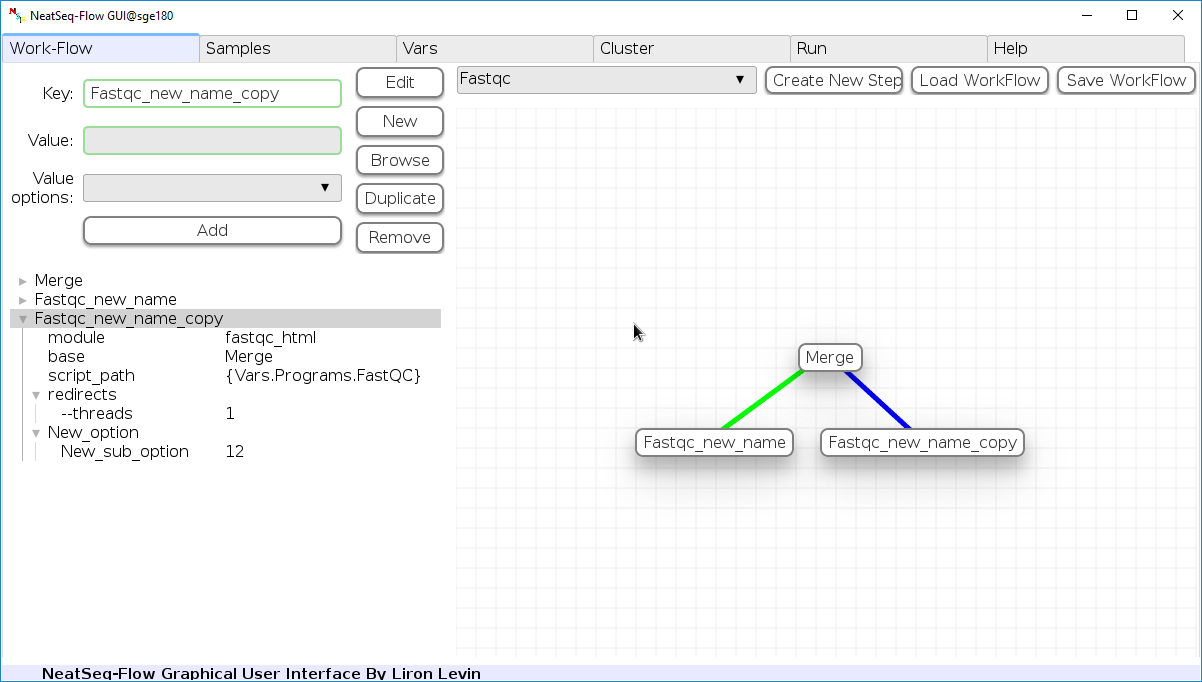
Duplicate field or step:
Click on the step’s name (to duplicate the step) or on a step’s option name (to duplicate the option and it’s sub fields)
Click the ‘Duplicate’ button
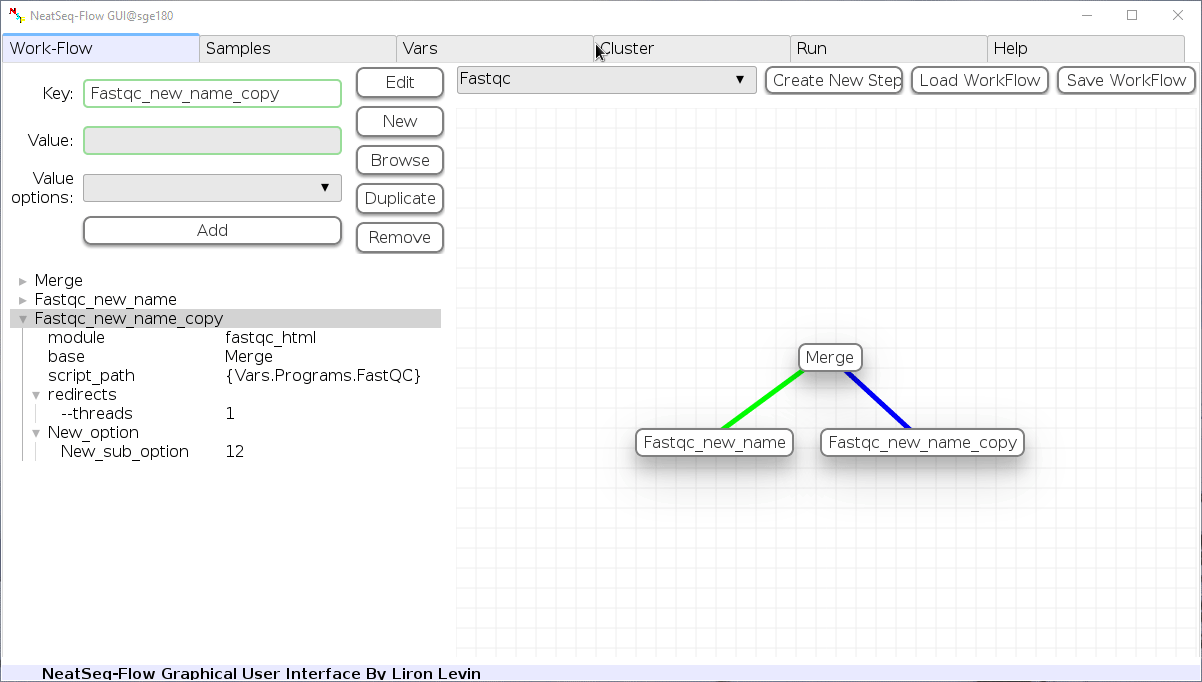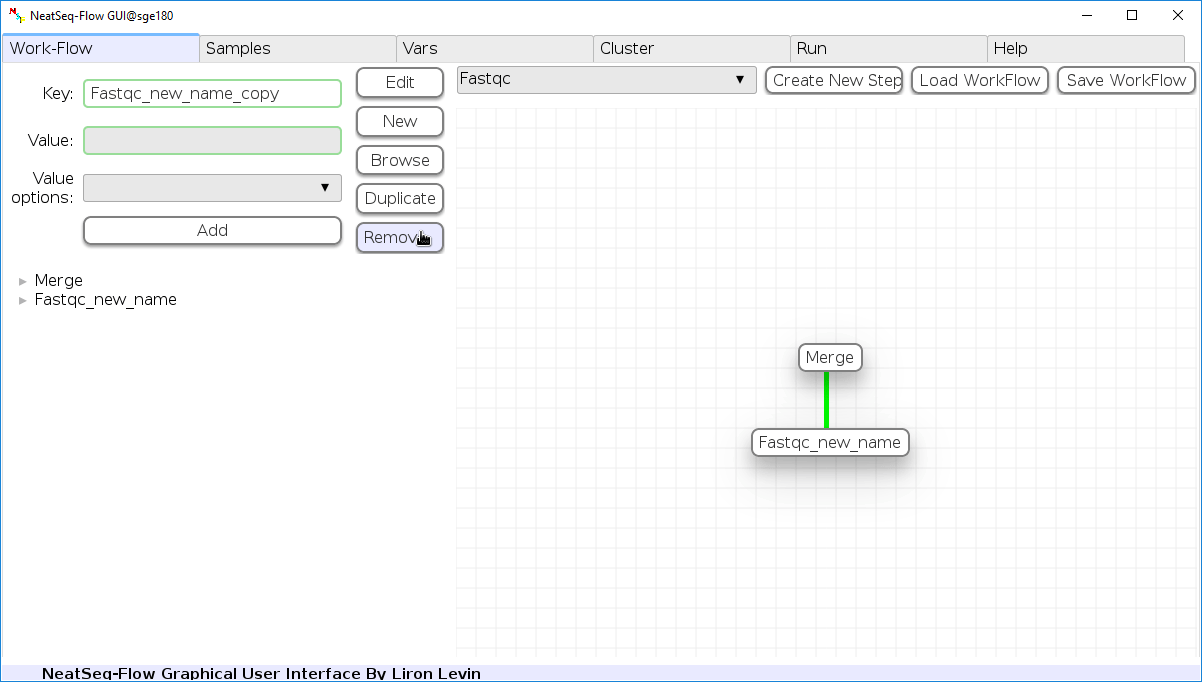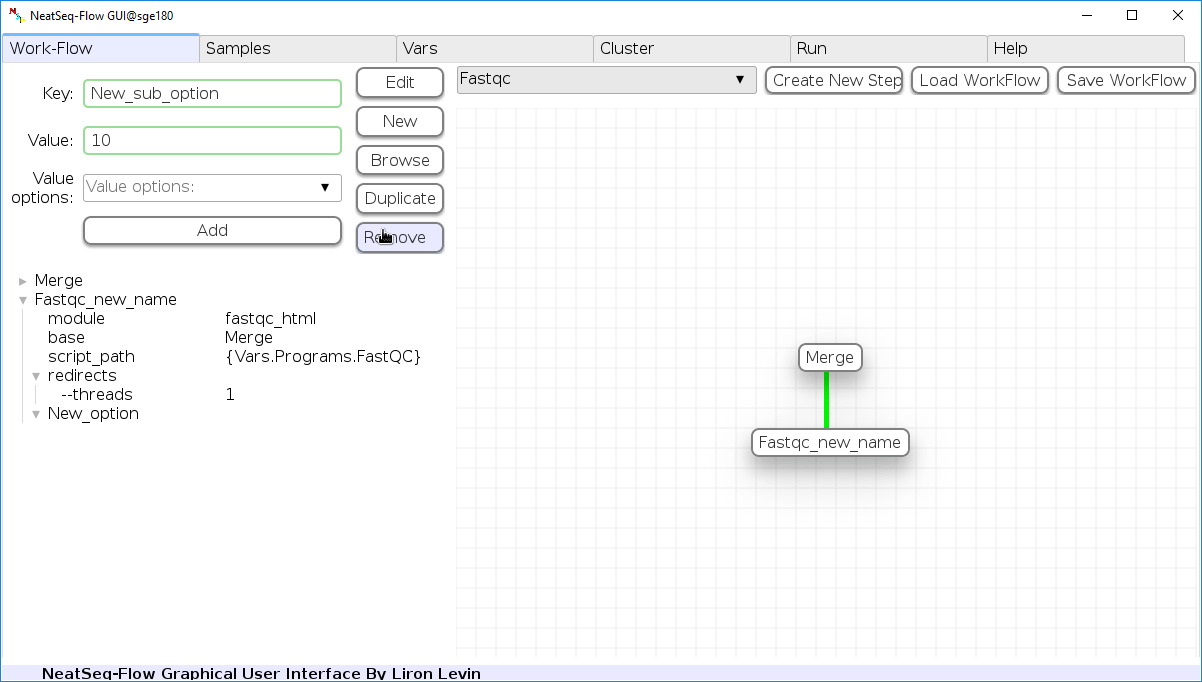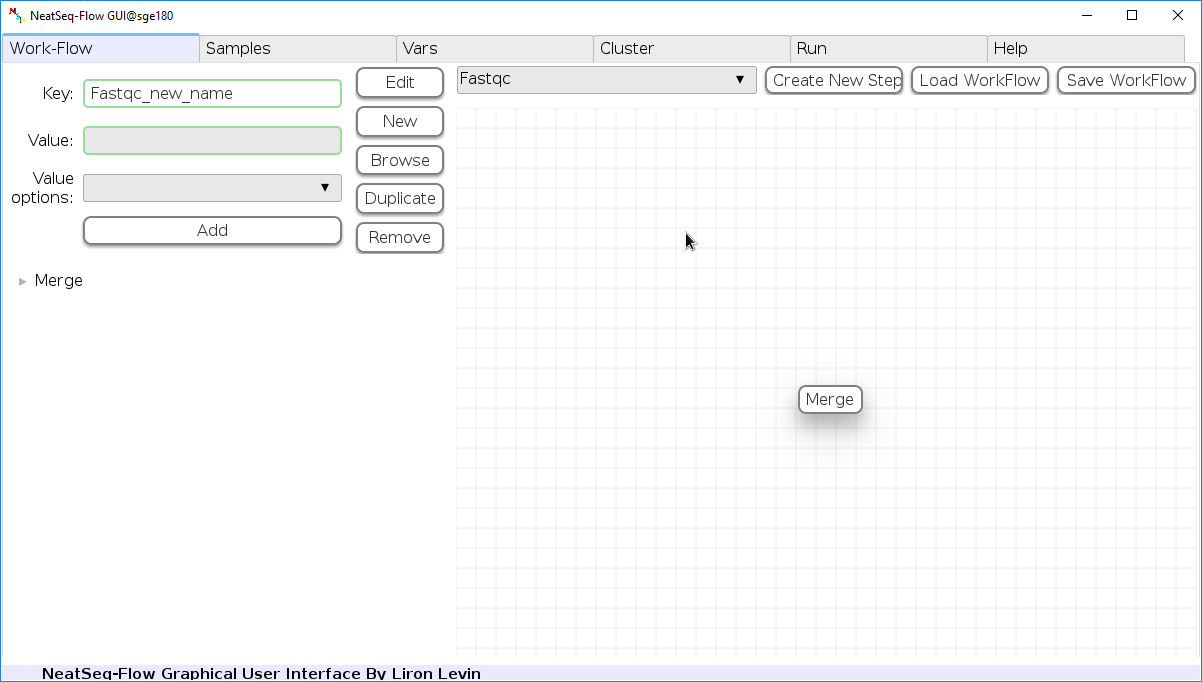
Remove field or step:
Click on the step’s name (to remove the step) or on a step’s option name (to remove the option and it’s sub fields)
In the ‘Vars’ Tab choose a variable name to edit, change the key or value and then click on the ‘Edit’ button.
Create New Variable:
You can create new variable by clicking on some existing variable name and then click the ‘New Field’ button.
You can create new sub variable by clicking on the existing variable to which you want to create a sub variable and then click the ‘New Sub Field’ button.
It is possible to indicate the Project Directory to generate the Work-Flow’s scripts in using the -d option (the default is to use the Current Working Directory )
It is possible to see all NeatSeq-Flow’s options by typing:
neatseq_flow.py-h
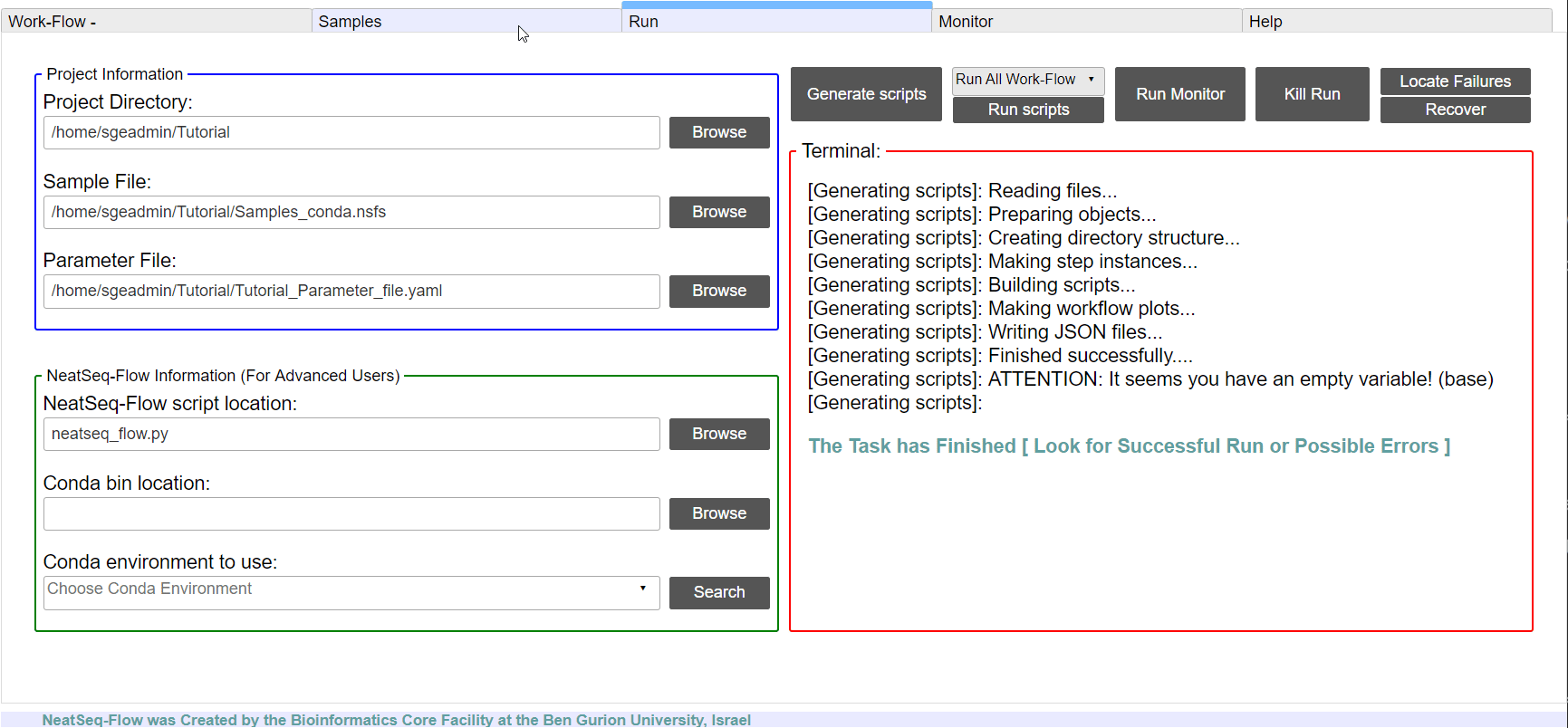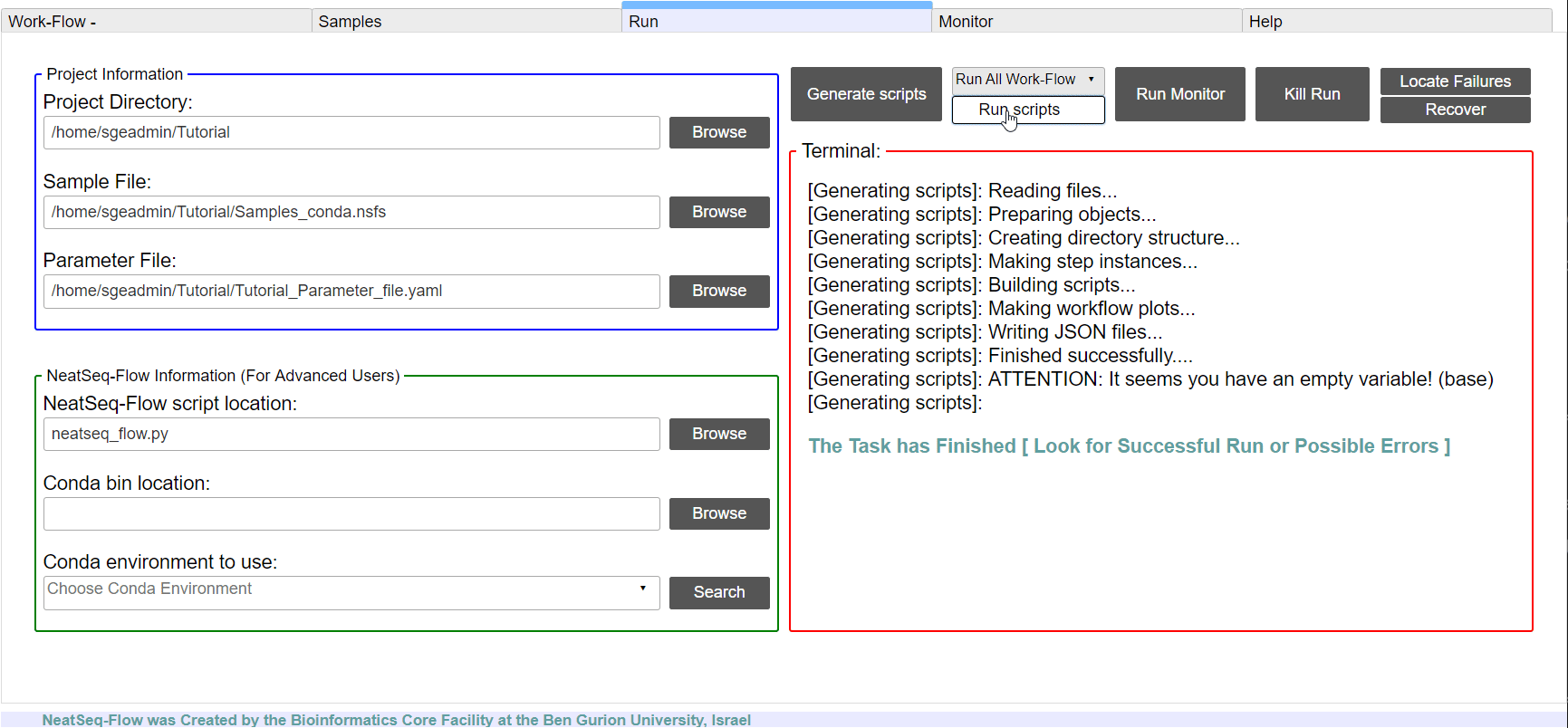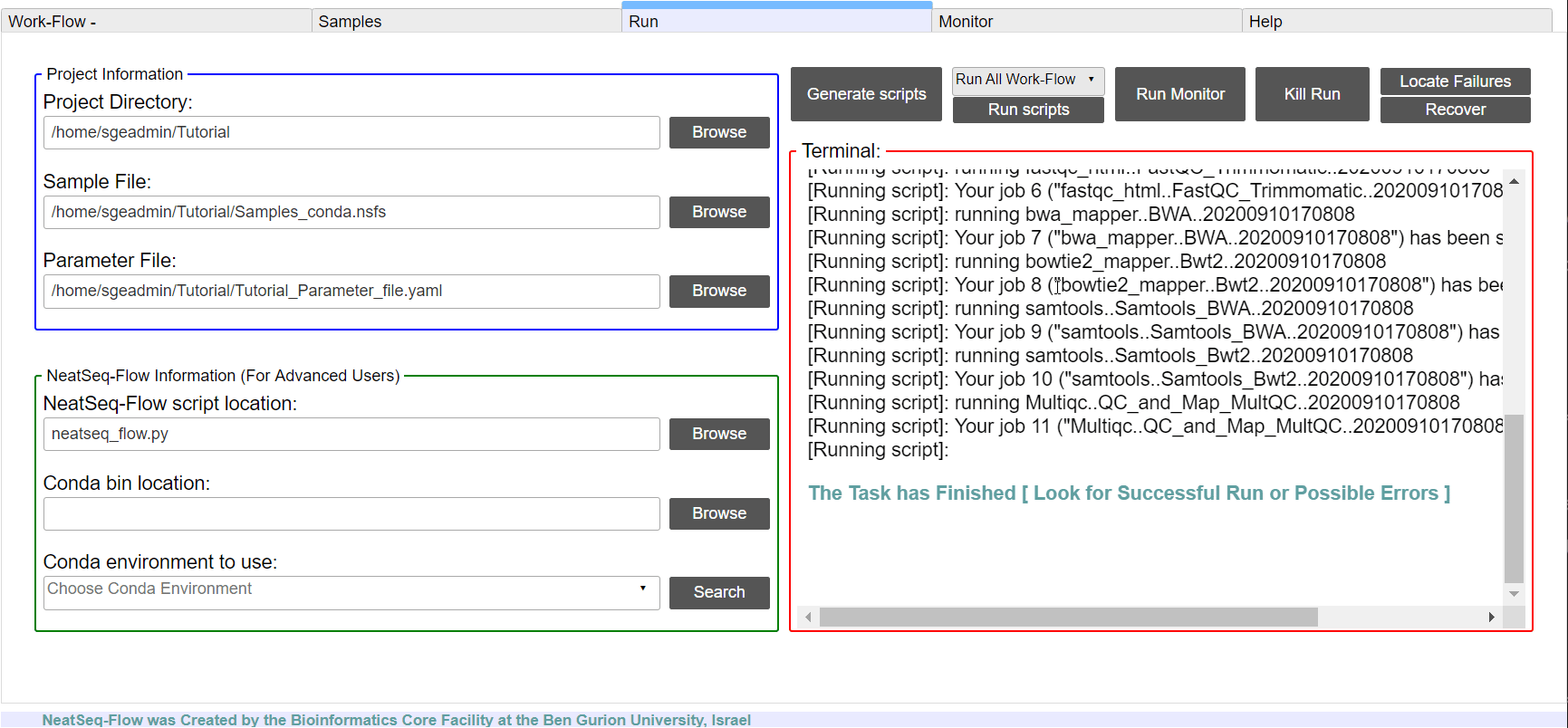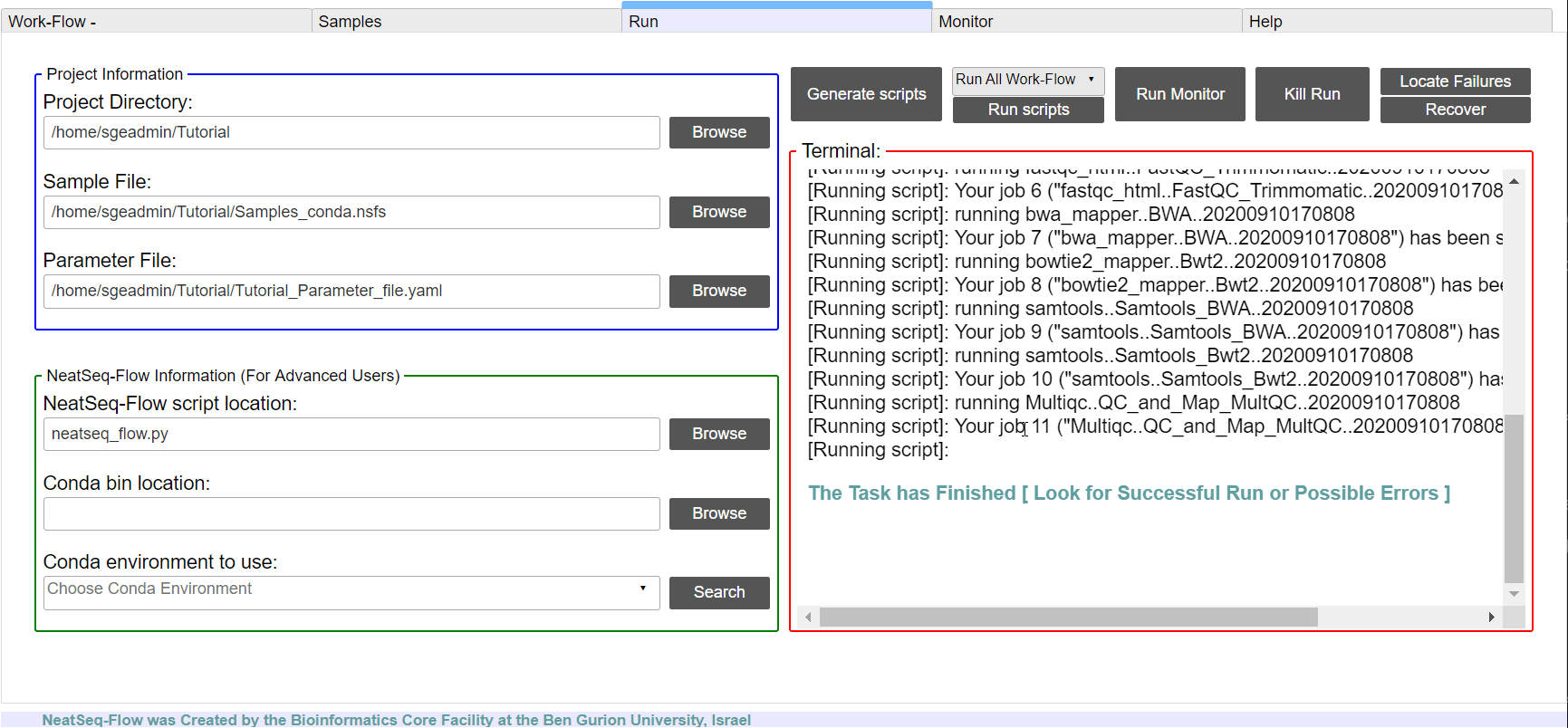
Run the Work-Flow by typing in the command line:
bashscripts/00.workflow.commands.sh1>null&
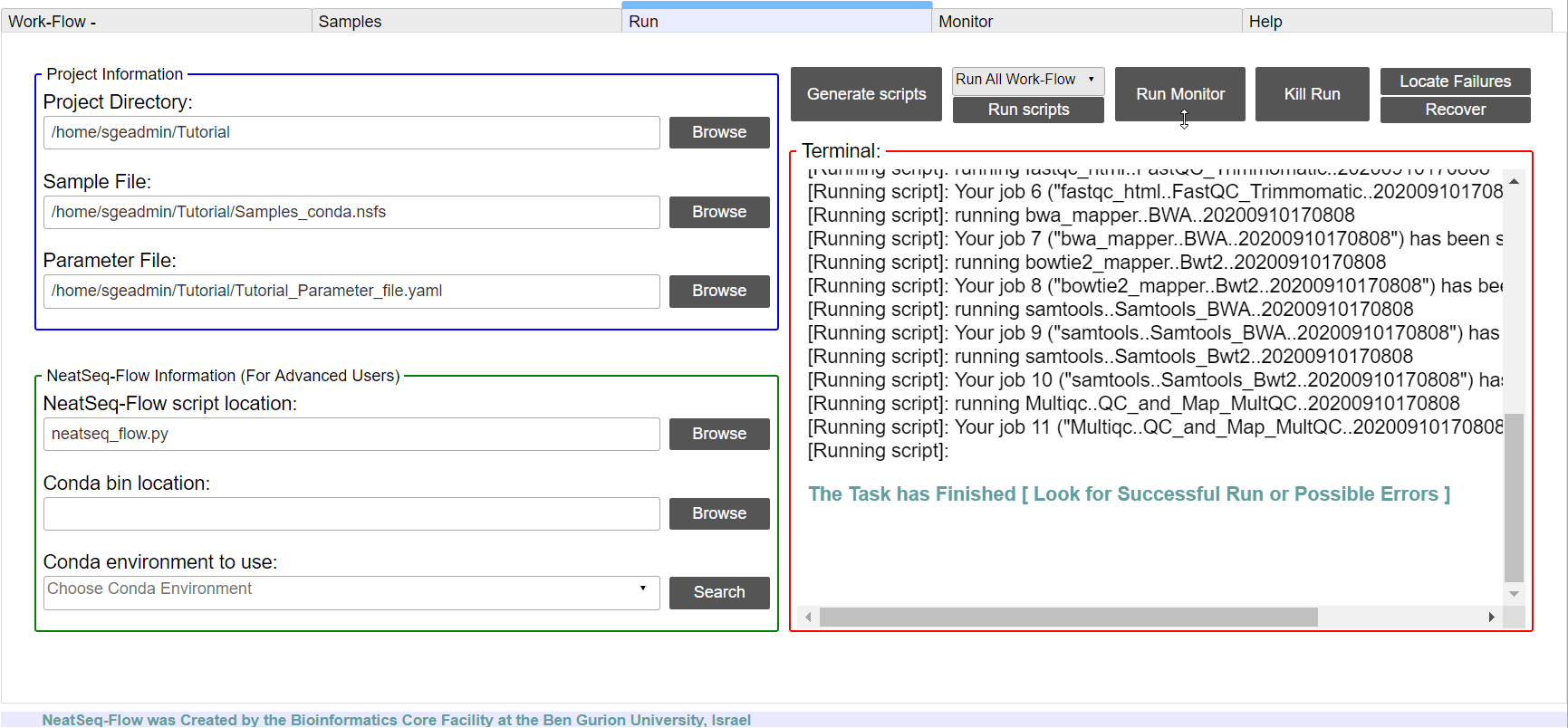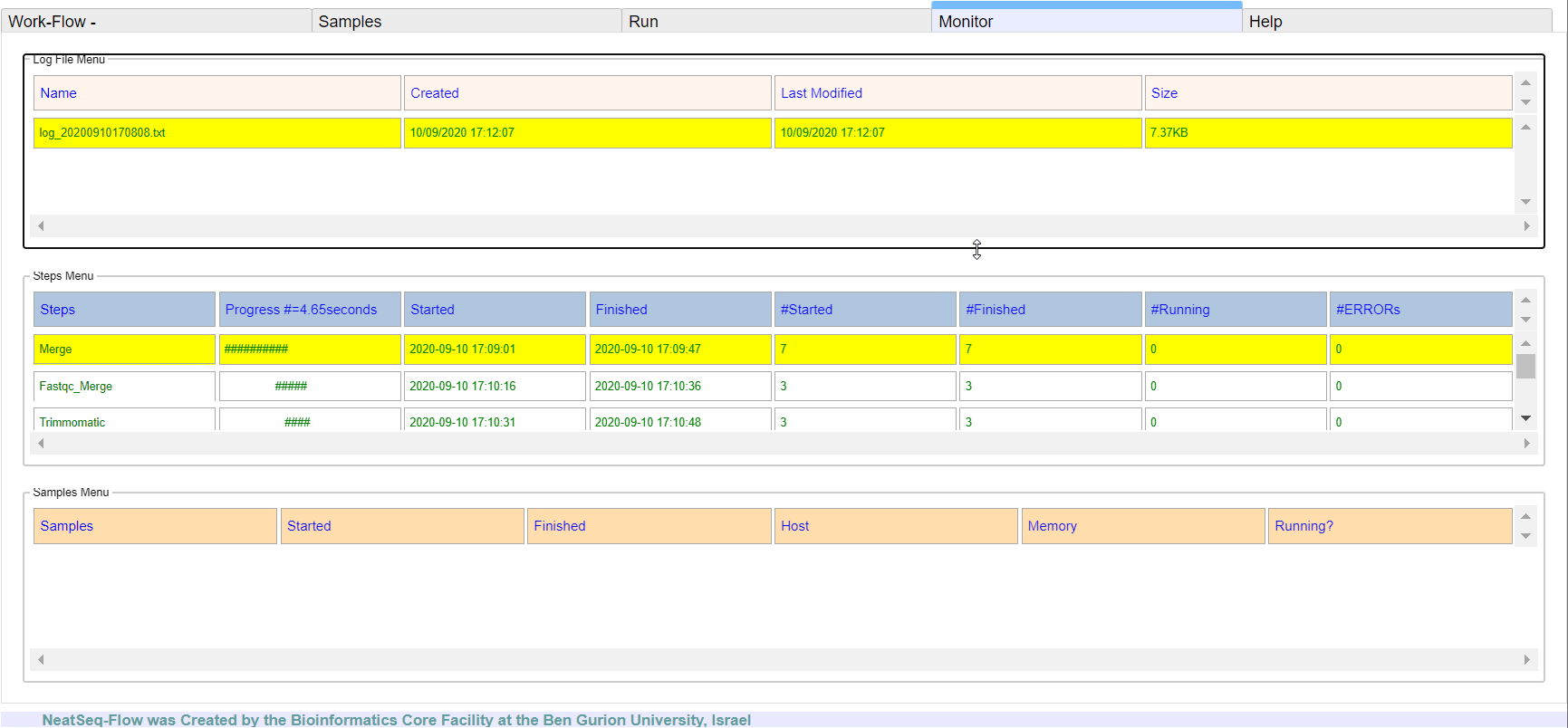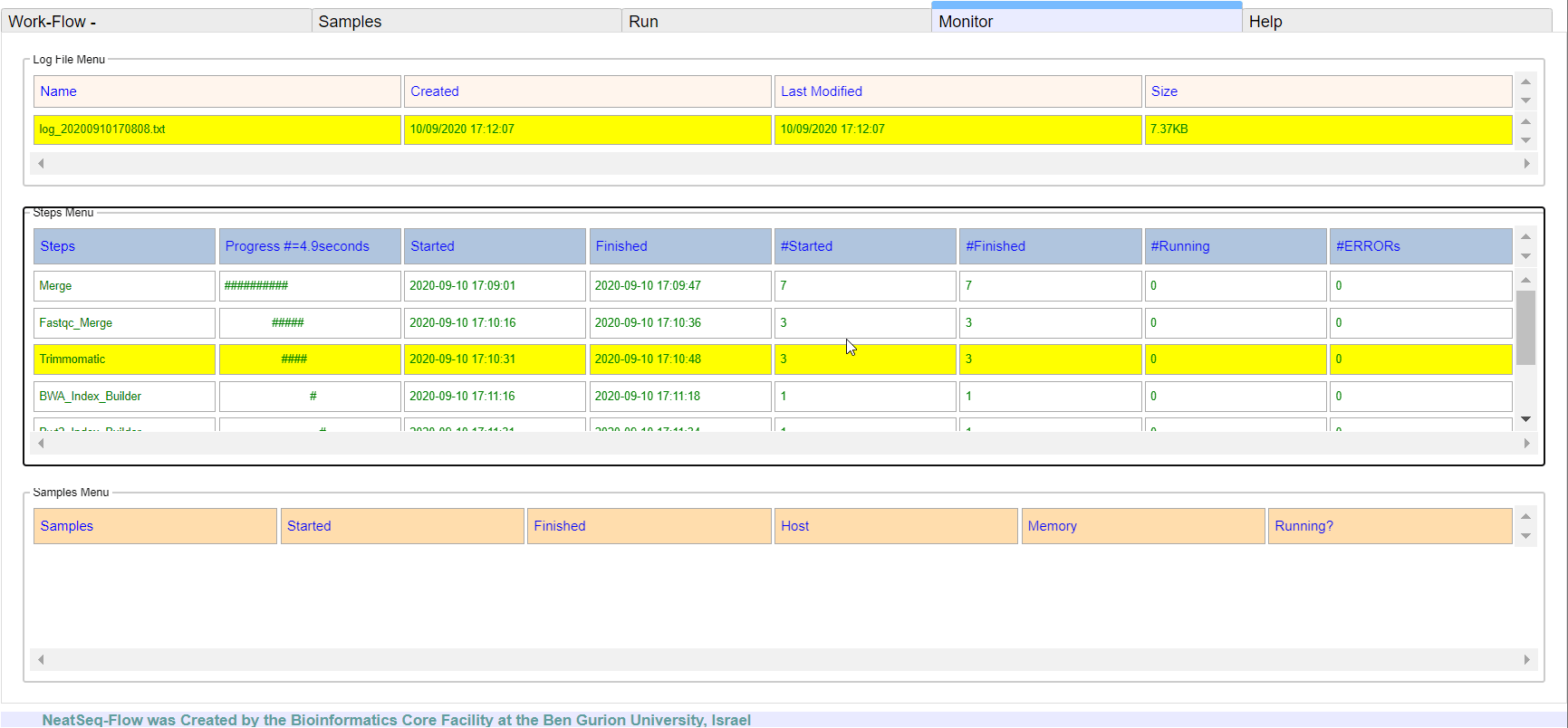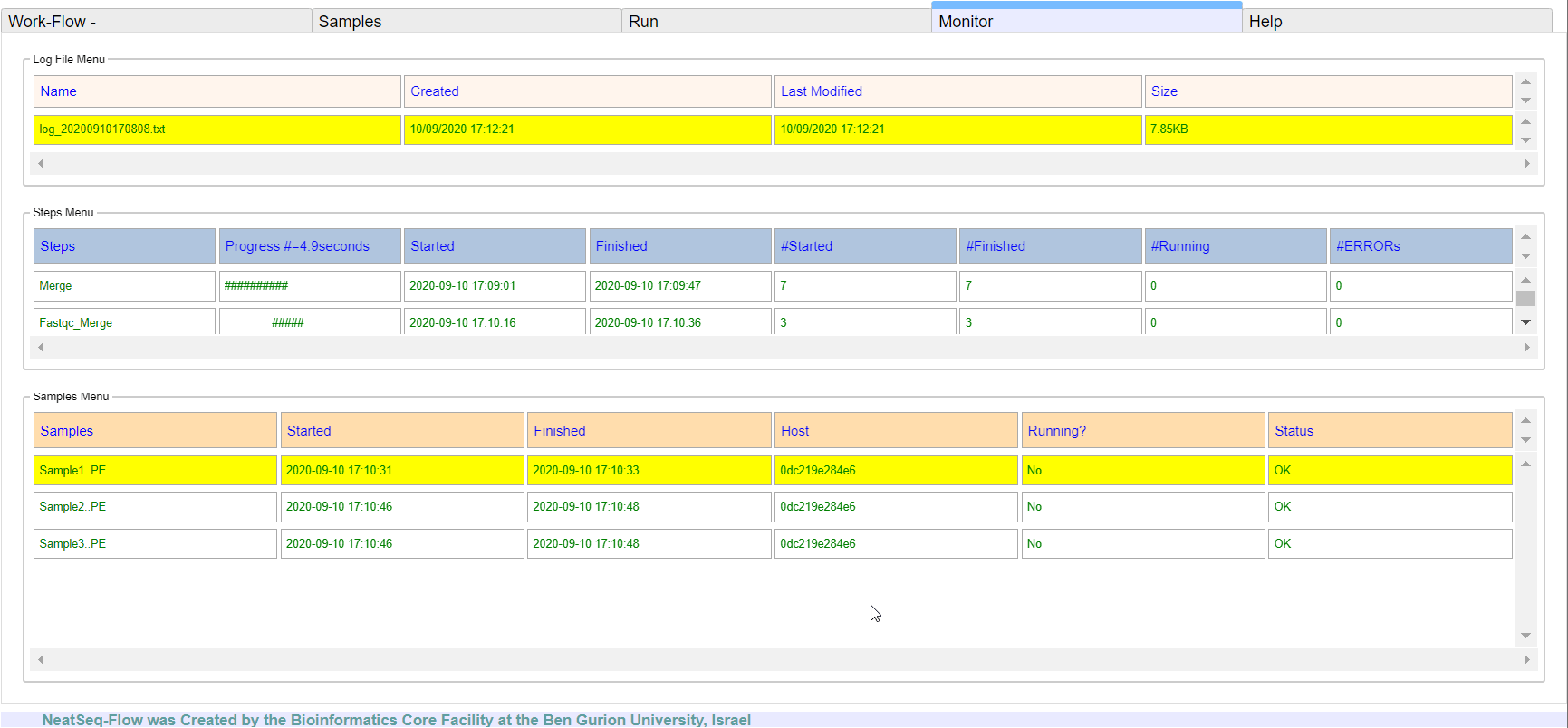
Run the Work-Flow monitor by typing in the command line:
neatseq_flow_monitor.py
Note
It is possible to terminate the current run by typing: