Transcriptome assembly and annotation with Trinity
- Author
Menachem Sklarz
- Affiliation
Bioinformatics Core Facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
Module categories
A pipeline for RNA-seq analysis using Trinity.
The workflow is built to run Trinity’s sample data, which consists of stranded, paired-end reads. You should modify the workflow steps to suit your data.
The workflow assembles a transcriptome with Trinity and then runs align_and_estimate_abundance.pl and abundance_estimates_to_matrix.pl to map the reads to the trascriptome and create normalized counts tables. The tables are used to select a representative transcript per gene, by expression.
Finally, the workflow uses Trinotate to annotate the resulting transcriptome.
Preparatory steps
If you want to use Trinotate, you must create and define the Trinotate databases 1.
If you already have the Trinotate databases downloaded and setup, you can set the paths to the databases in the
databasessubsection of theVarssection in the parameter file.See section Quick start with conda below for creating the databases with a conda installation.
If you want to use BUSCO:
Download a template config file with the following command and edit is as necessary. In the parameter file, set
Vars.paths.BUSCO_cfgto the full path to the config file.curl -L -o config.ini https://gitlab.com/ezlab/busco/raw/master/config/config.ini.default
Set the
Vars.databases.BUSCOvariable to the URL of the BUSCO dataset to use. Choose a URL from this list.Alternatively, download the lineage of interest, unzip it, and pass the path to the
BUSCOmodule via the--lineageredirect parameter.
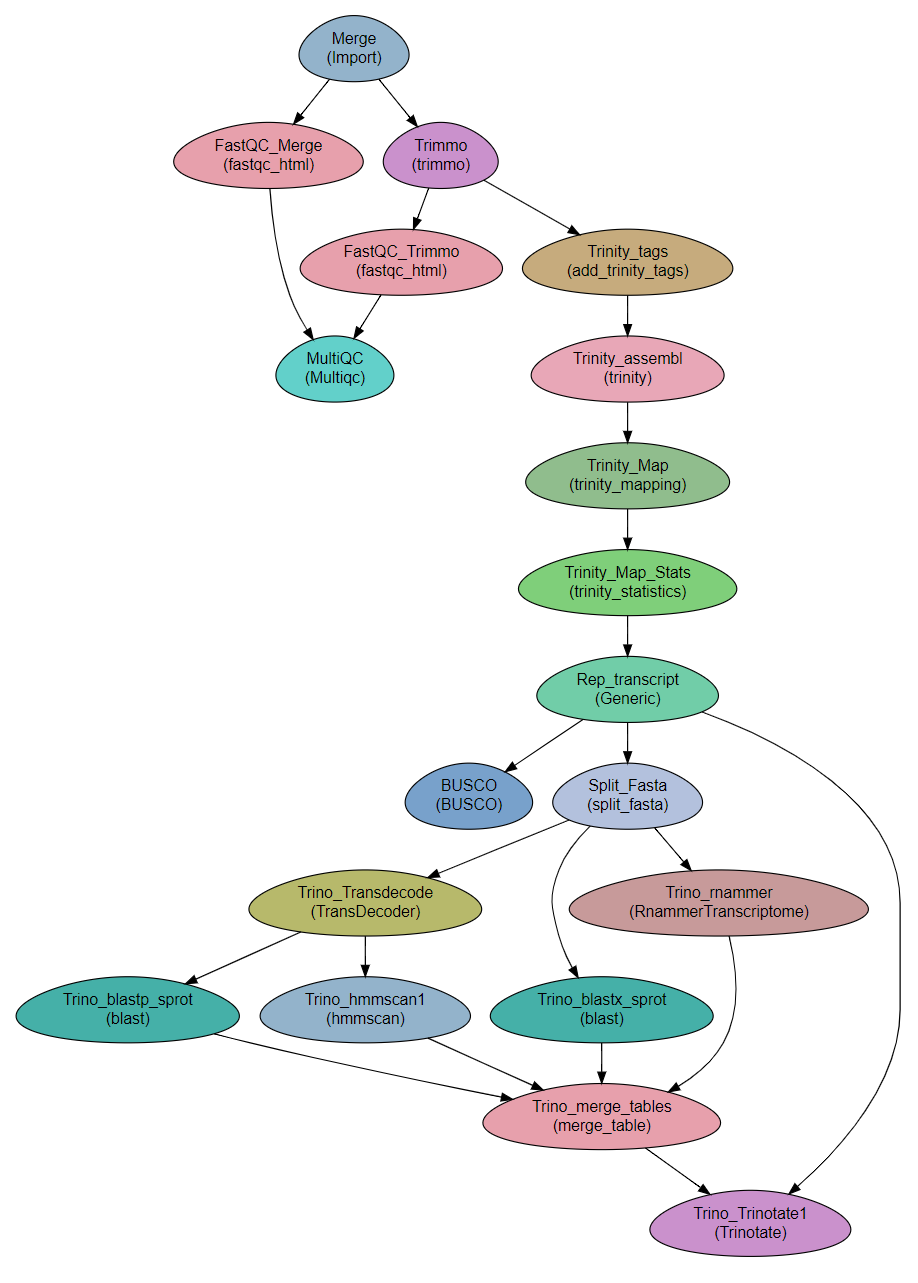
Steps
Importing and QC
Merge: Concatenating the read files into single files per direction
FastQC_Merge, Trimmo, FastQC_Trimmo and MultiQC: QC on the reads: FastQC and trimmomatic. Depending on the quality of the reads, trimmomatic` might not be required.
Trinity
Trinity_tags: Adding tags required by trinity to the read titles (/1 and /2 for F and R. See Running-Trinity. 2
Trinity_assembl: Running
Trinity. In this workflow, Trinity is executed on a single computer. Trinity can be configured to run on a cluster. The configuration file is set in theSGE_Trinity_confvariable in theVarssection. 3Trinity_Map: Mapping of the reads is performed with
trinity_mappingmodule.Trinity_Map_Stats: Creating statistical tables is performed with
trinity_statisticsmodule.Rep_transcript: Uses filter_low_expr_transcripts.pl to keep only a single transcript per gene.
BUSCO: Run BUSCO on the transcripts file. See Preparatory steps above for setting up BUSCO in the parameter file.
Trinotate
Split_Fasta: Splits the fasta file of transcripts for parallelization. From this step onwards, analysis is performed on subsets of the transcriptome. Recombining the results is done in step Trino_merge_tables.
Trino_blastx_sprot: Runs blastx against swissprot with the transcript sequences.
Trino_Transdecode: Finds coding sequences in the transcripts and produces predicted protein sequences.
Trino_blastp_sprot: Runs blastp against swissprot with the translated transcript sequences.
Trino_hmmscan1: Runs hmmscan against PFAM-A database with the translated transcript sequences.
Trino_rnammer: Runs RNAMMER to predict rRNA sequences in the transcripts.
Note
RNAMMER has to be set up in a special way. See here: https://github.com/Trinotate/Trinotate.github.io/wiki/Software-installation-and-data-required#rnammer-free-academic-download.
Trino_merge_tables: Merges the tables produced in the previous steps for the transcript subsamples.
Trino_Trinotate1: Read the tables and produce the final annotation file.
- 1
See instructions here: https://github.com/Trinotate/Trinotate.github.io/wiki/Software-installation-and-data-required#2-sequence-databases-required.
- 2
For the Trinity example described below, this step can be tagged with
SKIP, since the reads names already include tags.- 3
Trinity uses hpc_cmds_GridRunner for executing on grids. See their wiki for correct configuration.
Workflow Schema
Requires
fastq files. Paired end or single-end.
Programs required
Tip
See section Quick start with conda for installing all the programs with conda.
Example of Sample File
Title RNA_seq_denovo
#SampleID Type Path lane
Sample1 Forward /path/to/Sample1_F1.fastq.gz 1
Sample1 Forward /path/to/Sample1_F2.fastq.gz 2
Sample1 Reverse /path/to/Sample1_R1.fastq.gz 1
Sample1 Reverse /path/to/Sample1_R2.fastq.gz 2
Sample2 Forward /path/to/Sample2_F1.fastq.gz 1
Sample2 Reverse /path/to/Sample2_R1.fastq.gz 1
Sample2 Forward /path/to/Sample2_F2.fastq.gz 2
Sample2 Reverse /path/to/Sample2_R2.fastq.gz 2
Download
The workflow file is available here or with the following command:
curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/Workflows/RNA_seq_Trinity.yaml
Quick start with conda
For easy setup of the workflow, including a sample dataset, use the following instructions for complete installation with conda:
Attention
rnammer is not available with CONDA. To use it, you will have to install it and modify it following the instructions here.
Download the conda environment definition file:
You can
download the RNA_seq_Trinity_conda.yaml filehere, or programatically with:curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/docs/source/_extra/RNA_seq_Trinity_conda.yaml
Create and activate a conda environment with all the required programs:
conda env create -f RNA_seq_Trinity_conda.yaml source activate RNA_trinityGet the raw data from Trinity:
mkdir 00.Raw_reads cp $CONDA_PREFIX/opt/trinity-2.8.4/Docker/test_data/reads.right.fq.gz 00.Raw_reads/ cp $CONDA_PREFIX/opt/trinity-2.8.4/Docker/test_data/reads.left.fq.gz 00.Raw_reads/
Get
the sample filewith:curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/docs/source/_extra/RNA_seq_Trinity_samples.nsfs
Modify the paths in the sample file to the correct full paths.
Tip
If the raw read directory (00.Raw_data) and the sample file are in the same path, you can set the paths with the following sed command:
sed -i s+/path/to/+$PWD/+ RNA_seq_Trinity_samples.nsfs
Get
the parameter filewith:curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/Workflows/RNA_seq_Trinity.yaml
In the conda definitions (line 46), set
base:to the path to the conda installation which you used to install the environment.You can get the path by executing the following command:
echo $CONDA_EXE | sed -e 's/\/bin\/conda$//g'
If you want to use Trinotate, create a directory for the required databases (this step takes some time to complete):
mkdir Trinotate_dbs; Build_Trinotate_Boilerplate_SQLite_db.pl Trinotate_dbs/Trinotate mv uniprot_sprot.* Trinotate_dbs/ mv Pfam-A.hmm.gz Trinotate_dbs/ cd Trinotate_dbs/ makeblastdb -in uniprot_sprot.pep -dbtype prot gunzip Pfam-A.hmm.gz hmmpress Pfam-A.hmm cd - .. Attention:: If you already have the Trinotate databases downloaded and setup, you do not have to do the last steps. You can set the paths to the databases in the ``databases`` subsection of the ``Vars`` section in the parameter file.
If you want to use BUSCO:
Download a template config file with the following command and edit is as necessary:
curl -L -o config.ini https://gitlab.com/ezlab/busco/raw/master/config/config.ini.default
Set the Vars.databases.BUSCO variable to the URL or the BUSCO dataset to use. Choose a URL from this list: https://busco.ezlab.org/frame_wget.html.
Deactivate the RNA_trinity environment:
conda deactivate