NeatSeq-Flow Tutorial Workflow
- Author
Menachem Sklarz and Liron Levin
- Affiliation
Bioinformatics Core Facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
Module categories
This tutorial describes how to create and execute the workflow described in the NeatSeq-Flow manuscript (Article on BioRXiv).
See NeatSeq-Flow Tutorial for detailed instructions for quick installation of the tutorial workflow with conda.
The example workflow receives FASTQ files and the sequenced genome of a bacteria. It then performs:
Quality testing and trimming of the raw sequence reads (paired- or single-end).
Alignment (“mapping”) of the reads to a reference genome using two different programs.
Sorting the samples’ BAM files as final results.
Creation of a report on reads and mapping quality.
Workflow Schema
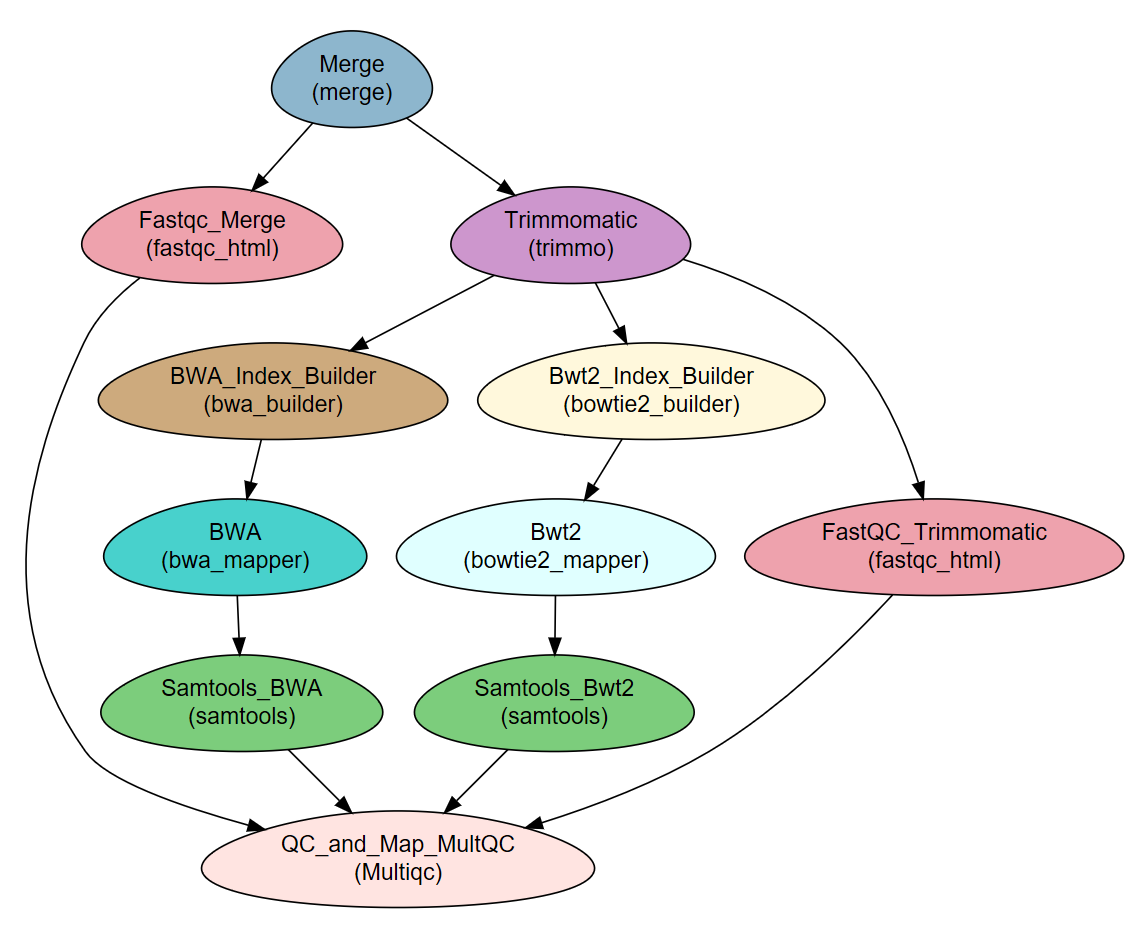
Steps
Step |
Module |
Program |
|---|---|---|
Merge |
merge |
|
Fastqc_Merge |
fastqc_html |
fastqc |
Trimmomatic |
trimmo |
trimmomatic |
FastQC_Trimmomatic |
fastqc_html |
fastqc |
BWA_Index_Builder |
bwa_builder |
bwa |
BWA |
bwa_mapper |
bwa |
Bwt2_Index_Builder |
bowtie2_builder |
bowtie2 |
Bwt2 |
bowtie2_mapper |
bowtie2 |
Samtools_BWA |
samtools |
samtools |
Samtools_Bwt2 |
samtools |
samtools |
QC_and_Map_MultQC |
Multiqc |
MultiQC |
Required data
This WF requires samples with fastq file(s) (paired or single) and a reference genome in fasta format.
Programs required
fastqc
trimmomatic
multiqc
samtools=1.3
BWA
bowtie2
Example of Sample File
Title Example_WF_From_the_manuscript
#Type Path
Nucleotide /path/to/Reference_genome.fasta
#SampleID Type Path
Sample1 Forward /path/to/Sample1.F.fastq.gz
Sample1 Reverse /path/to/Sample1.R.fastq.gz
Sample2 Forward /path/to/Sample2.F.fastq.gz
Sample2 Reverse /path/to/Sample2.R.fastq.gz
Sample3 Forward /path/to/Sample3.F.fastq.gz
Sample3 Reverse /path/to/Sample3.R.fastq.gz
Download
curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-tutorial/master/Samples_conda.nsfs
curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-tutorial/master/Example_WF_conda_env.yaml