Microbiome analysis using QIIME¶
| Author: | Menachem Sklarz |
|---|---|
| Affiliation: | Bioinformatics Core Facility |
| Organization: | National Institute of Biotechnology in the Negev, Ben Gurion University. |
Module categories
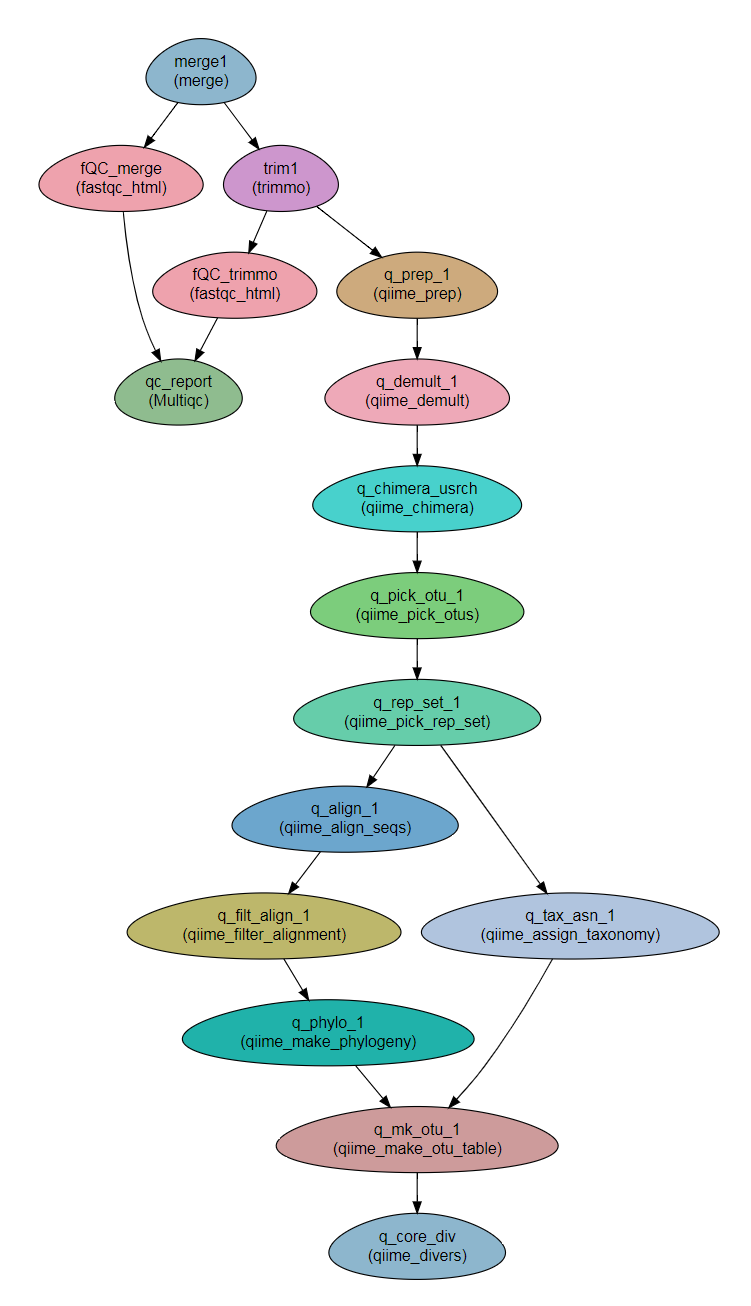
A workflow for executing a complete QIIME analysis (based on QIIME 1.9.. A new workflow for QIIME2 is also available)
Developed as part of a study led by Prof. Jacob Moran-Gilad.
Steps¶
- Read preparation:
- merge
- trimmomatic - For cleaning the reads
- FastQC - Checking the quality of the reads
- MultiQC
- Optional read joining with join_paired_ends.py (module
qiime_prep) - Creating symbolic links to files so that demultiplication can recognise the sample names from the link names.
- Further QA and concatenating all sequences into a single fasta file with
qiime_demult. - Identifying chimeric sequences (
qiime_chimeramodule`)
- Analysis (denovo OTU picking)
- OTU picking
- selecting representative sequences
- Aligning to reference and filtering the alignment
- Assigning taxonomic lineage to representative sequences
- Creating phylogenetic tree
- Creating BIOM table
- Computing core diversity analyses on the BIOM table.
- Optional
- You can filter out particular samples or OTUs with modules
qiime_filter_samples_from_otu_tableandqiime_filter_otus, respectively. - You can sort the BIOM table samples by a specific field of the mapping file with the
qiime_sort_otu_tablemodule
- You can filter out particular samples or OTUs with modules
Example of Sample File¶
Title Metagenomics
#SampleID Type Path lane
Sample1 Forward /path/to/Sample1_F1.fastq.gz 1
Sample1 Forward /path/to/Sample1_F2.fastq.gz 2
Sample1 Reverse /path/to/Sample1_R1.fastq.gz 1
Sample1 Reverse /path/to/Sample1_R2.fastq.gz 2
Sample2 Forward /path/to/Sample2_F1.fastq.gz 1
Sample2 Reverse /path/to/Sample2_R1.fastq.gz 1
Sample2 Forward /path/to/Sample2_F2.fastq.gz 2
Sample2 Reverse /path/to/Sample2_R2.fastq.gz 2