RNA-Seq using a reference genome¶
| Author: | Liron Levin |
|---|---|
| Affiliation: | Bioinformatics Core Facility |
| Organization: | National Institute of Biotechnology in the Negev, Ben Gurion University. |
Page Contents:
Note
In order to use this Work-Flow first:
- Install NeatSeq-Flow using conda
- Make sure that conda is in your PATH.
Steps¶
- Merge Decompression and Concatenation (IF NEADED) of read files into single files per direction.
- FastQC_Merge Quality tests on the original reads using FastQC.
- MultiQC_pre_trim Quality report on the original reads using MultiQC.
- Trim_Galore Reads trimming using Trim_Galore.
- FastQC_Trim_Galore Quality tests on reads after trimming using FastQC.
- RSEM_Genome indexing of the reference genome, mapping of the post trimming reads and count data creation.
- MultiQC_post_trim Quality report on the trimmed reads and mapping information using MultiQC.
Workflow Schema¶
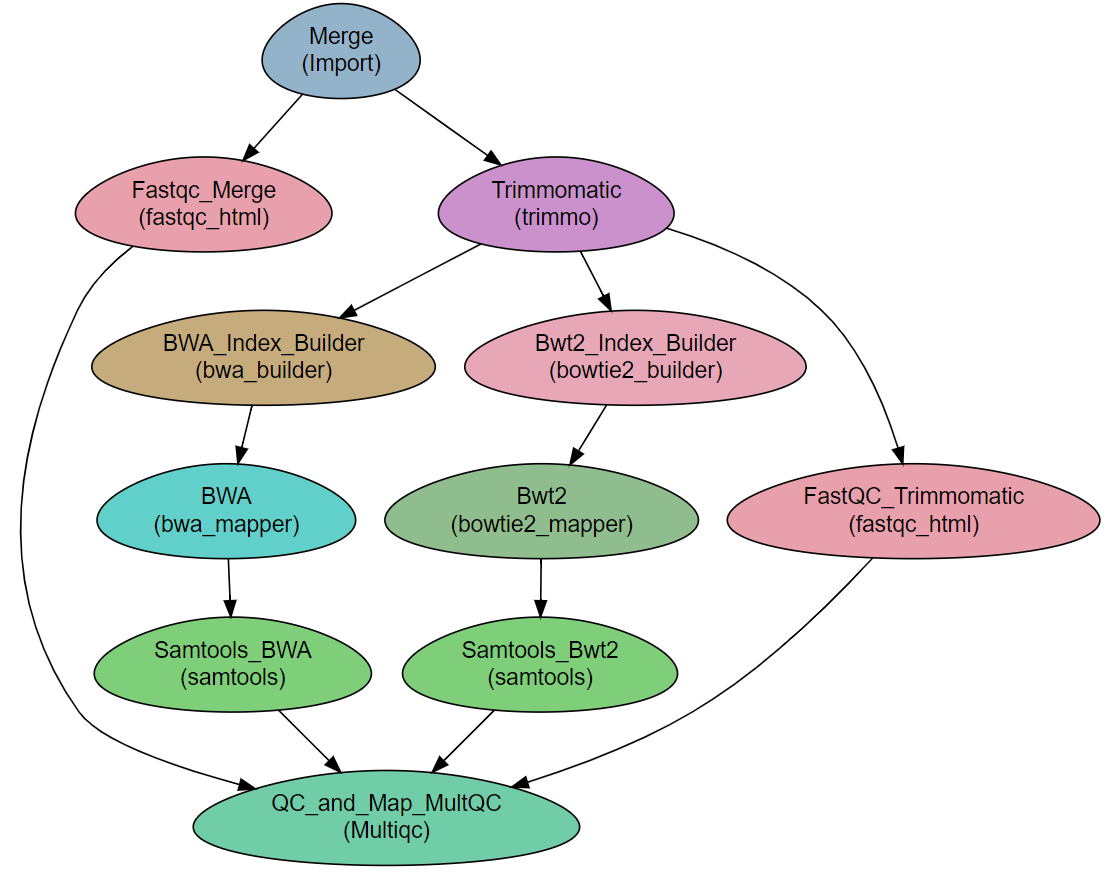
Note
It is possible to add a DeSeq2 step for Differential Expression, Clustering and Functional Analyses. For more information see the DESeq2 Tutorial
Requires¶
- Paired end or single-end reads fastq files. .
- A reference genome in fasta format
- An annotation file in gtf format
Programs required¶
Note
The programs are installed as part of the installation process using CONDA.
Example of Sample File¶
Create a tab-delimited sample file. It should look as follows:
Title RNA_seq
#SampleID Type Path
Sample1 Forward /path/to/Sample1_F1.fastq.gz
Sample1 Forward /path/to/Sample1_F2.fastq.gz
Sample1 Reverse /path/to/Sample1_R1.fastq.gz
Sample1 Reverse /path/to/Sample1_R2.fastq.gz
Sample2 Forward /path/to/Sample2_F1.fastq.gz
Sample2 Reverse /path/to/Sample2_R1.fastq.gz
Sample2 Forward /path/to/Sample2_F2.fastq.gz
Sample2 Reverse /path/to/Sample2_R2.fastq.gz
Note
You can edit the file in excel but make sure to save it in tab-delimited format. See this section of the manual for a full description of the sample file format.
Quick start with conda¶
Install all the required programs in to a conda environment:
Download the
RNASeq conda environment installer file:curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/docs/source/_extra/RNASeq_env_install.yamlCreate the RNASeq conda environment:
conda env create -f RNASeq_env_install.yaml
Download the Work-Flow’s Parameter file:
curl https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/Workflows/RNASeq_STAR.yaml > RNASeq.yamlcurl https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/Workflows/RNASeq_Bowtie2.yaml > RNASeq.yaml
Activate the NeatSeq_Flow conda environment:
bash source activate NeatSeq_Flow
- Edit the “Vars” section in the Work-Flow’s Parameter file:
Specify the location of the gtf and reference genome files
Note
It is recommended to use the NeatSeq-Flow GUI in order to:
- Edit the Work-Flow’s Parameter file
- Create a Samples file
- Generate and run the Work-Flow’s scripts.
NeatSeq_Flow_GUI.py
Learn more about How to use NeatSeq-Flow GUI
Alternatively, It is possible to use a text editor.
Generate the scripts by typing in the command line:
neatseq_flow.py -s Samples_file.nsfs -p RNASeq.yaml
Run the Work-Flow by typing in the command line:
bash scripts/00.workflow.commands.sh 1> null &
Run the Work-Flow monitor by typing in the command line:
neatseq_flow_monitor.py