ChIP-seq workflow
- Author
Menachem Sklarz
- Affiliation
Bioinformatics Core Facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
Module categories
This workflow automates a standard ChIP-seq analysis.
Note
This workflow is based on a workflow kindly provided by Dr. Dena Leshkowitz of the Life Sciences Core Facilities, Weizmann Institute of Science.
Warning
The ChIP-seq workflow is in active development.
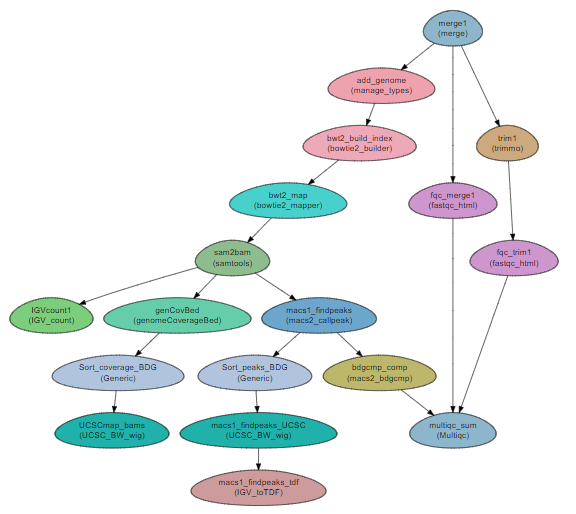
Steps:
Preparation and QA: a. Merging the reads into a single file per sample (
merge). b. QC with fastqc (fastqc_html) c. Trimming with trimmomatic (trimmo) d. QC on trimmed reads with fastqc e. Adding the genome and the GTF file to the project (manage_types).Mapping a. Creating a bowtie2 index for the genome (
bowtie2_builder) a. Mapping the reads to the reference genome with bowtie2 (bowtie2_mapper) b. Conversion to sorted BAM with samtools (samtools) c. Sorting by name for bedGraphToBigWig with theGenericmodule. c. Converting to UCSC and IGV format (genomeCoverageBed,UCSC_BW_wigandIGV_count)Finding ChIP peaks a. Peak calling is performed with macs2 callpeak (
macs2_callpeak) b. Further analysis of the peaks is done with macs2 bdgcmp (macs2_bdgcmp)
Attention
In this workflow, we added the genome and GTF file via the parameter file. It is possible to add them via the sample file, as well.
Workflow Schema
Requires
fastq files, either paired-end or single-end.
A sample to control mapping (see Example sample lines below)
Programs required
Tip
All programs can be installed with CODDA. See section Quick start with conda
Example of Sample File
Title ChIP_project
#SampleID Type Path lane
Sample1 Forward /path/to/Sample1_F1.fastq.gz 1
Sample1 Forward /path/to/Sample1_F2.fastq.gz 2
Sample1 Reverse /path/to/Sample1_R1.fastq.gz 1
Sample1 Reverse /path/to/Sample1_R2.fastq.gz 2
Sample2 Forward /path/to/Sample2_F1.fastq.gz 1
Sample2 Reverse /path/to/Sample2_R1.fastq.gz 1
Sample2 Forward /path/to/Sample2_F2.fastq.gz 2
Sample2 Reverse /path/to/Sample2_R2.fastq.gz 2
Sample_Control Sample1:Sample2
Download
The workflow file is available here
Quick start with conda
For easy setup of the workflow with CONDA, use the following instructions:
Create and activate a conda environment with all the required programs:
curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/docs/source/Workflow_docs/ChIP_seq_conda.yaml conda env create -n ChIP_seq_WF -f ChIP_seq_conda.yaml source activate ChIP_seq_WF
Create a sample file. It should look like the file shown in Example of Sample File. Don’t forget to replace the sample names and file paths:
Warning
Make sure the file is TAB-delimited!
Tip
To get the full path to a file, use the following command:
readlink -f <filename>
Get the parameter file with:
curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/Workflows/ChIP_seq.yaml
Run the workflow:
Activate the NeatSeq-Flow conda environment. (See Installing NeatSeq-Flow)
Execute the script generator and run the workflow. (See Running NeatSeq-Flow.)