Microbiome analysis using QIIME2
- Author
Menachem Sklarz
- Affiliation
Bioinformatics Core Facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
Attention
This workflow is in active development!
Captive and Wild Atlantic Salmon project
Background
This workflow is based on the data described in Structural and compositional mismatch between captive and wild Atlantic salmon (Salmo salar) parrs gut microbiota highlights the relevance of integrating molecular ecology for management and conservation methods 1.
The data for the workflow is available on datadryad.
The workflow demonstrates executing qiime2 on a set of illumina paired-end reads.
The data for the workflow includes the raw reads and a metadata file. Obtaining the files will be demostrated in a later section.
The workflow also downloads a classifier object. This file is not included in the sample file as it is not specific to the project at hand. NeatSeq-Flow can also be programmed to train your own classifier but this option is beyond the scope of this workflow.
Attention
Please note this workflow is a demonstration of how to use the qiime2 with NeatSeq-Flow. Please make sure you go over the steps and parameters to make sure it suits your needs!
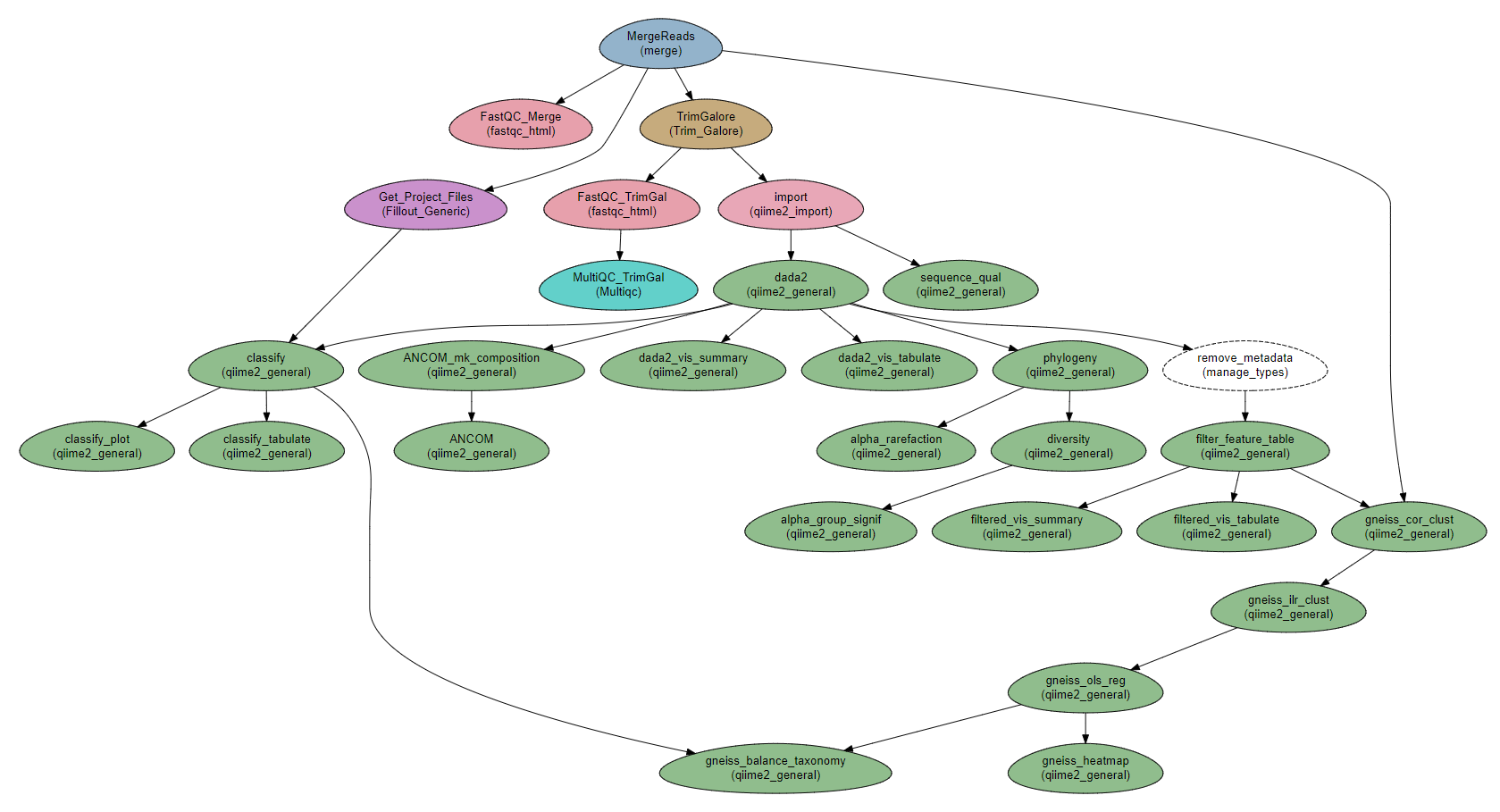
Steps
MergeReads: Get the data and files from the sample file.
FastQC_Merge, TrimGalore, FastQC_TrimGal, and MultiQC_TrimGal: QC on the reads: FastQC and Trim Galore!. Depending on the quality of the reads, TrimGalore` might not be required.
Get_Project_Files: Download and import the classifier file into the workflow.
import: import sequence data into a QIIME2 artifact
sequence_qual: Create quality report for the sequences.
dada2: dada2 and visualization.
dada2_vis_summary:
dada2_vis_tabulate:
remove_metadata and filter_feature_table: Filter low-expression features from the feature table (have to first remove metadata slot, to avoid filtering by metadata)
filtered_vis_summary and filtered_vis_tabulate: Visualization of the filtered table.
Tip
From this step onwards, if you want to use the filtered feature table, base your steps on filter_feature_table.
phylogeny: Building a phylogenetic tree
diversity: Core diversity analysis
alpha_rarefaction: Creating α-rarefaction curves.
alpha_group_signif: alpha groups differences based on Faith’s diversity index.
Classification:
Using the classifier downloaded in step Get_Project_Files.
Tip
You can add steps in the workflow to train your own classifier, but that is beyond the scope of this workflow.
classify: Classify the reads.
classify_tabulate and classify_plot: Visualization of the classification.
gneiss_*: Steps for executing the gneiss analysis
ANCOM*: Steps for executing the ANCOM analysis
Workflow Schema
Requires
Raw reads for the analysis can be downloaded as follows (Note: The downloaded directory is 5.5 GB!):
wget https://datadryad.org/stash/downloads/file_stream/67648 tar zxv 16S_reads_salmo_salar_V3_V4_gut_microbiota.tar.gz
Get
the salmon sample filewith:curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/docs/source/_extra/QIIME2/qiime2.samples.salmon.nsfs
Get the
qiime2-formatted metadata filewith:curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/docs/source/_extra/QIIME2/qiime2.metadata.salmon.tsv
Modify the paths in the sample file to the correct full paths.
Tip
If the raw read directory (16S_reads_salmo_salar_V3_V4_gut_microbiota), the metadata file and the sample file are in the same path, you can set the paths with the following sed commands:
sed -i s+/path/to/+$PWD/16S_reads_salmo_salar_V3_V4_gut_microbiota/+ qiime2.samples.salmon.nsfs
sed -i s+qiime2.metadata.salmon.tsv+$PWD/qiime2.metadata.salmon.tsv+ qiime2.samples.salmon.nsfs
Programs required
QIIME2, version 2019.4, installed with conda as described here.
fastqc,multiqc,TrimGalore!andcutadaptwhich are not included in the qiime2 environment. All of these can be installed in a separate conda environment with:curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/docs/source/_extra/QC_conda.yaml conda env create -f QC_conda.yaml
You can also
download the file from here
Download
The workflow file is available for download with the following command:
curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/Workflows/qiime2.analysis.salmon.yaml
Attention
The following instructions assume the NeatSeq-Flow, qiime2 and QC environments were installed with the same conda version!
After downloading the parameter file, set the conda env in the Vars section to the name of the qiime environment you installed above, typically something like qiime2-2018.11.
Execute NeatSeq-Flow
Execute NeatSeq-Flow with the sample and parameters files downloaded above:
source activate NeatSeq_Flow export CONDA_BASE=$(conda info --root) neatseq_flow.py -s qiime2.samples.salmon.nsfs -p qiime2.analysis.salmon.yaml
The Moving Pictures tutorial
A workflow for executing the Moving Windows tutorial with QIIME2.
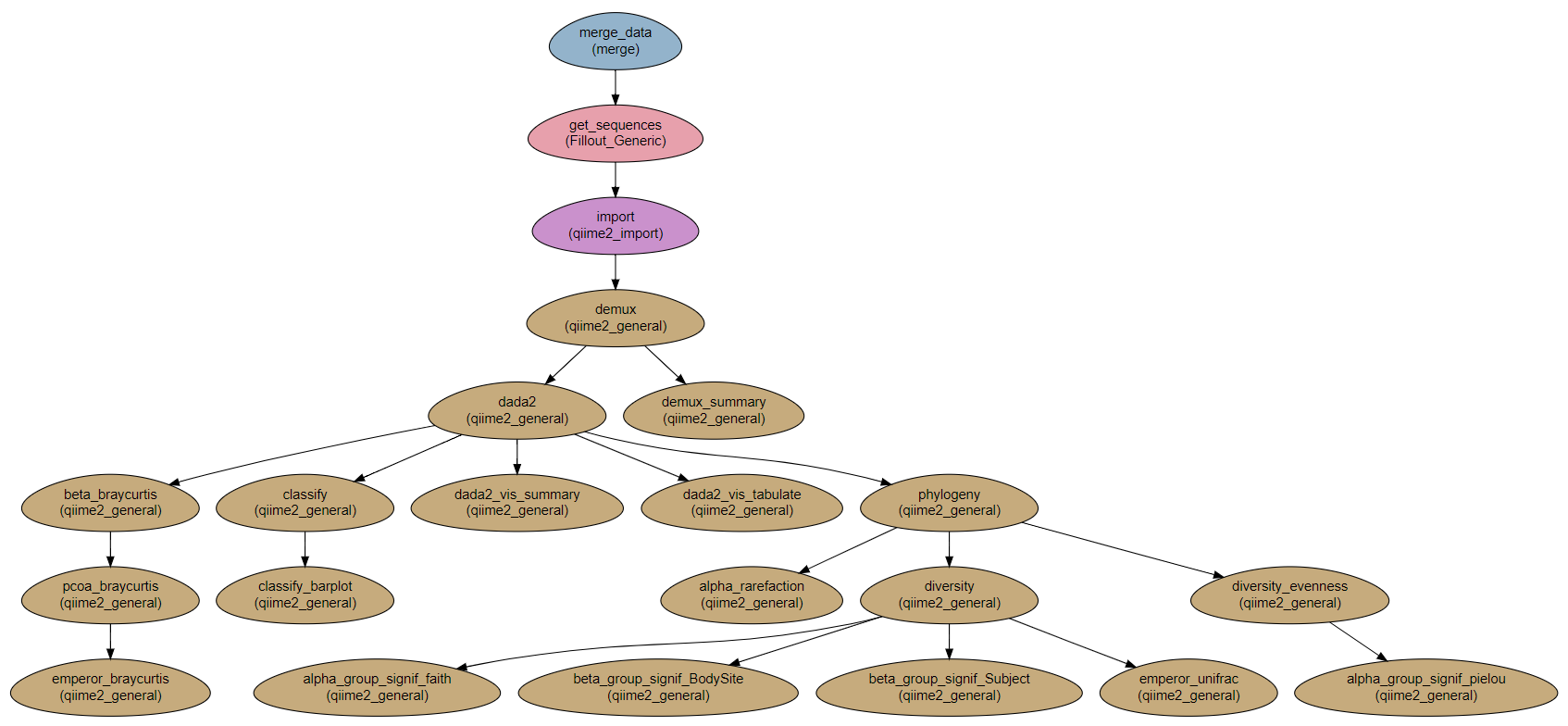
Steps:
Merge_data: Get the data and files from the sample file.
Get_sequences: Download the sequences from the internet
import: import sequence data into a QIIME2 artifact
demux: Demultiplex.
demux_summary: Show statistics of demultiplexed data
dada2: dada2 and visualization.
dada2_vis_summary:
dada2_vis_tabulate:
phylogeny: Building a phylogenetic tree
diversity: Core diversity analysis
diversity_evenness: Calculating Pielou’s evenness index.
Comparing alpha and beta groups differences.
alpha_group_signif_faith: alpha groups differences based on Faith’s diversity index.
alpha_group_signif_pielou: alpha groups differences based on Pielou’s evenness index.
beta_group_signif_BodySite: beta groups differences based on site in body.
beta_group_signif_Subject: beta groups differences based on subject.
Creating emperor visualizations.
emperor_unifrac: Emperor visualization based on UniFrac index.
beta_braycurtis, pcoa_braycurtis and emperor_braycurtis: Emperor visualization based on Bray-Curtis index.
alpha_rarefaction: Creating α-rarefaction curves.
Taxonomy:
classify: taxonomic classification
classify_barplot: taxonomy visualization with barplots.
Workflow Schema
Requires
No requirements. All files are downloaded by the workflow.
Programs required
QIIME2, version 2018.11, installed with conda as described here.
Attention
Download the parameter file in the link below and set the conda path in line 10 to the location of your conda installation, not including bin. e.g., if using the default location of miniconda, the path should be $HOME/miniconda2.
Download
The workflow and sample files are available for download with the following commands:
curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/Workflows/qiime2_MovingPic_fullAuto.params.yaml
curl -LO https://raw.githubusercontent.com/bioinfo-core-BGU/neatseq-flow-modules/master/Workflows/qiime2_MovingPic_fullAuto.samples.nsfs