Microbiome analysis using QIIME
- Author
Menachem Sklarz
- Affiliation
Bioinformatics Core Facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
Module categories
A workflow for executing a complete QIIME analysis (based on QIIME 1.9.. A new workflow for QIIME2 is also available)
Developed as part of a study led by Prof. Jacob Moran-Gilad.
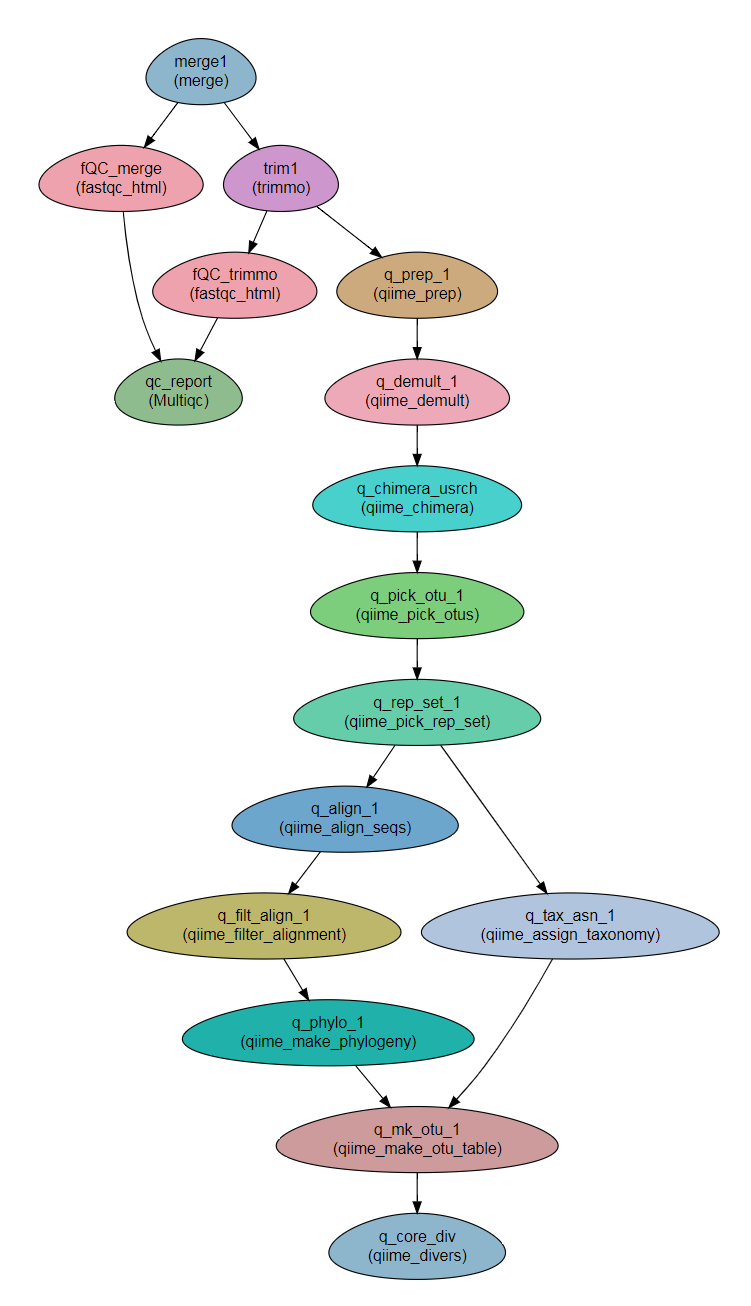
Steps
- Read preparation:
merge
trimmomatic - For cleaning the reads
FastQC - Checking the quality of the reads
MultiQC
Optional read joining with join_paired_ends.py (module
qiime_prep)Creating symbolic links to files so that demultiplication can recognise the sample names from the link names.
Further QA and concatenating all sequences into a single fasta file with
qiime_demult.Identifying chimeric sequences (
qiime_chimeramodule`)
- Analysis (denovo OTU picking)
OTU picking
selecting representative sequences
Aligning to reference and filtering the alignment
Assigning taxonomic lineage to representative sequences
Creating phylogenetic tree
Creating BIOM table
Computing core diversity analyses on the BIOM table.
- Optional
You can filter out particular samples or OTUs with modules
qiime_filter_samples_from_otu_tableandqiime_filter_otus, respectively.You can sort the BIOM table samples by a specific field of the mapping file with the
qiime_sort_otu_tablemodule
Workflow Schema
Requires
fastq files. Paired end or single-end.
Programs required
Example of Sample File
Title Metagenomics
#SampleID Type Path lane
Sample1 Forward /path/to/Sample1_F1.fastq.gz 1
Sample1 Forward /path/to/Sample1_F2.fastq.gz 2
Sample1 Reverse /path/to/Sample1_R1.fastq.gz 1
Sample1 Reverse /path/to/Sample1_R2.fastq.gz 2
Sample2 Forward /path/to/Sample2_F1.fastq.gz 1
Sample2 Reverse /path/to/Sample2_R1.fastq.gz 1
Sample2 Forward /path/to/Sample2_F2.fastq.gz 2
Sample2 Reverse /path/to/Sample2_R2.fastq.gz 2
Download
The workflow file is available here