Generic Modules
The generic modules, called Generic and Fillout_Generic, do not contain a definition of input and output file types, therefore the user has to specify the input and output file types in the parameter file.
Genericis simpler to use for defining most Linux programs, and has extra file type management capacities.Fillout_Genericcan incorporate more than one command per step, as well as cater to irregular program calls, such as calls including complex pipes; however, using it is slightly more complicated.
Modules included in this section
Generic
- Authors
Liron Levin
- Affiliation
Bioinformatics core facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
Short Description
A generic module that enables the user to design new modules that can handle most cases.
Requires
In this module the users define the required file types in the inputs section
Output
In this module the users define the output file types in the outputs section
The scope of the output file types is determinant by the module scope
Parameters that can be set
Parameter |
Values |
Comments |
|---|---|---|
scope |
sample/project |
The scope of this module could be sample/project, the default is by sample |
shell |
csh/bash |
Type of shell [csh OR bash]. bash is the default, only bash can be used in conda environment |
inputs_last |
The inputs arguments will be at the end of the command |
Example of usage and implementation of the generic module:
Attention
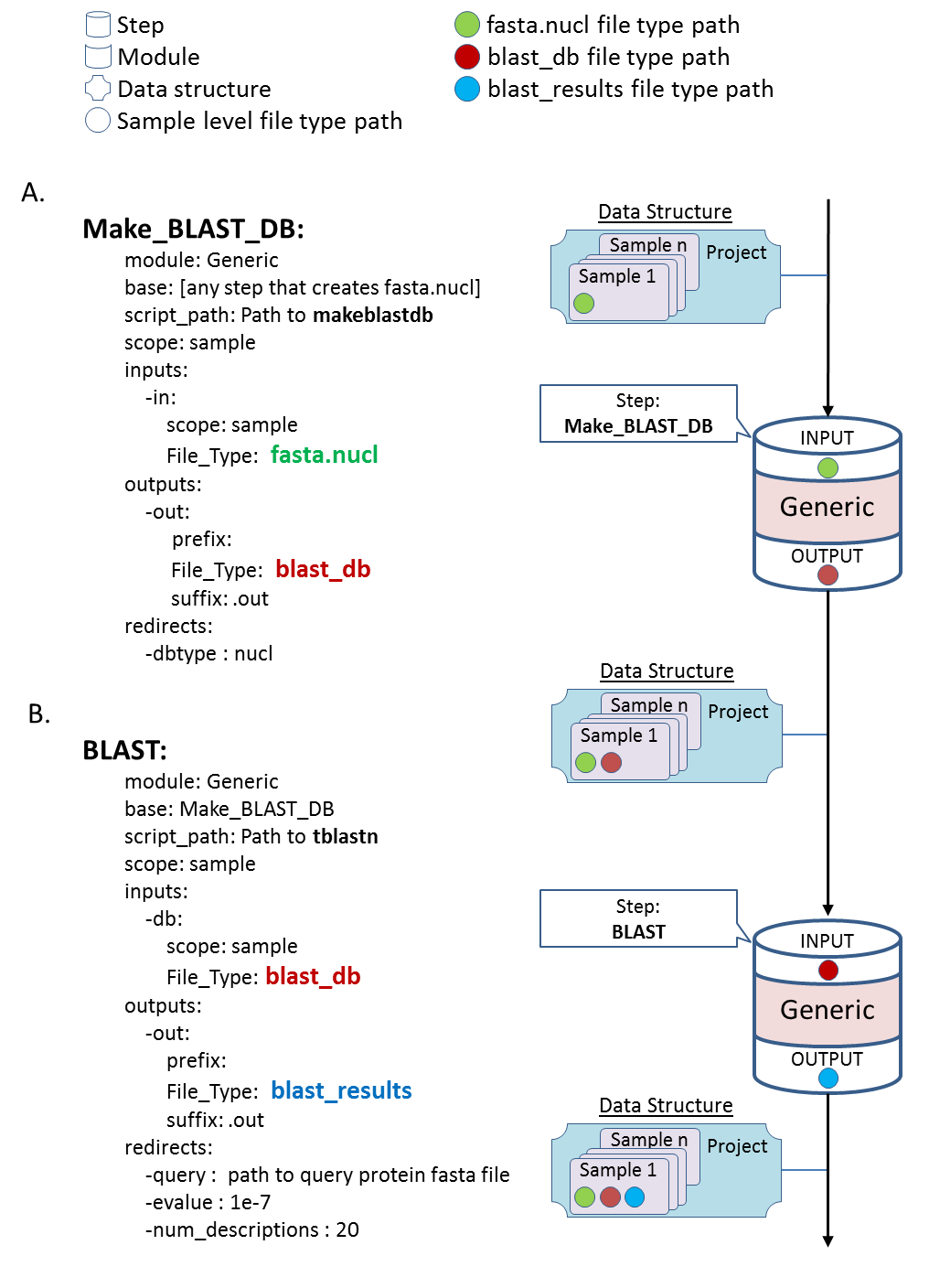
A generic module is used to generate a BLAST database for each sample and a subsequent generic step queries each database with sequences from an external FASTA file. This example is a typical use of BLAST in many biological scenarios such as searching for virulence/resistance genes (whose sequences are in the external FASTA file) in bacterial genomes
A. Calling a generic module to generate a BLAST database (using makeblastdb) from each sample. This step can be used after (base:) any step that creates a nucleotide FASTA file (File_Type: fasta.nucl), e.g. after merge (if the raw files are in nucleotide FASTA format) or after a de novo assembly step. The location of the BLAST database for each sample is saved as a blast_db file type (File_Type: blast_db) for downstream use. B. Calling a generic module which performs a BLAST search (tblastn) of an external query protein fasta file (-query : path to query protein fasta file) against the previously generated BLAST data base per sample. This step can be used after the Make_BLAST_DB step (base: Make_BLAST_DB). The user can pass additional parameters directly to the used program in the redirects section (e.g. –dbtype, –evalue, -num_descriptions etc.).
Lines for parameter file
Step_Name: # Name of this step
module: Generic # Name of module
base: # Name of the step [or list of names] to run after [must be after steps that generates the inputs File_Types]
script_path: # Main command for this module
scope: # The scope of this module could be sample/project, the default is by sample
shell: # Type of shell [csh OR bash]. bash is the default. only bash can be used in conda environment
arg_separator: # The separator between the arguments and values [The default is space].
inputs_last: # The inputs arguments will be at the end of the command. [The default is inputs arguments at the beginning of the command]
command_order: # The order of the command parts as string default 'redirects,inputs,outputs' ignored if inputs_last is set.
use_base_dir: # Use the base step directory as the output for this step, it is possible to specify the base to use.
cd: # Change current working directory to the output location.
no_sample_dir: # In Sample Scope: will NOT create a dedicated folder for each sample and the location of the base folder will be stored
# in a project level 'base_dir' File_Type
remove_subsamples: # Will remove subsamples created by previous steps (split_fasta for example)
subsamples_string: # A string to identify a subsample, all subsample will start with this string. [default: 'subsample']
inputs: # The inputs for this module
STR: # Input argument, e.g. -i, --input [could be also 'empty1', 'empty2'.. for no input argument string]
scope: # The scope of this input argument could be sample/project
# If the module scope is project and the argument scope is sample:
# all the samples inputs File_Types of this argument will be listed as: [input argument] [File_Type(sample#)] e.g. -i sample1.bam -i sample2.bam ...
File_Type: # The input File_Type could be any File_Type available from previous (in this branch) steps
# It is possible to indicate more then one File_Type separated by comma 'fastq.F,fastq.R'
base: # From which previous step to take the input File_Type. The default is the current step.
sep: # If the module scope is project and the argument scope is sample:
# All the samples inputs File_Types of this argument will be listed delimited by sep. e.g. [sep=,] -i sample1.bam,sample2.bam ...
# If more then one File_Type was specify the inputs File_Types of this argument will be listed delimited by sep.
prefix: # A prefix for this input argument file name
suffix: # A suffix for this input argument file name
use_dirname: # Use only the input Directory and add suffix for constant file name and prefix to add a string before the input Directory
del: # Delete the files in the input File_Type after the step ends [use to save space for large files you don't need downstream]
# Will generate empty file with the same name and a suffix of _DELETED
constant_value: # use a constant value instead of "File_Type".
# it is the same as the "redirects".
# use when the order of inputs are important!!
# use '{{sample_name}}' to be replace with the sample name (or project name in project scope)
# using the constant_value option will override all other input arguments!!!!!!
outputs: # The outputs for this module
STR: # Output argument, e.g. -o, --out , the scope of the output arguments is determinant by the module scope
# could be also 'empty1', 'empty2'.. for no output argument string OR 'No_run1', 'No_run2'.. for only entering the file information to output File_Type
File_Type: # The output File_Type could be any File_Type name for the current branch downstream work
# If the File_Type exists its content will be override for the current branch downstream work
prefix: # A prefix for this output argument file name
suffix: # A suffix for this output argument file name
# between prefix and suffix will be the sample name [in sample scope] or the project title [in project scope]
constant_file_name: # Use constant file name for this output argument [ignore prefix and suffix]
# If empty [''] will enter the output directory location
use_base_name: # use only the base name of the output file [ignored if constant_file_name is used]
copy_File_Types: # Transferring information between File_Types
STR: # Unique name for the transfer
source:
File_Type: # Copy the content of source File_Type to the target File_Type [copy from here]
scope: # Copy the source File_Type From this scope [if not specified the default is sample]
base: # The source step to copy the File_Type from (from previous steps). The default it the current step.
constant_value: # Use to transfer information outside of the 'File_Type' system to a File Type, will always be considered as project scope
# Using the constant_value option will override all other source arguments!!!!!!
target:
File_Type: # Copy the content of source File_Type to the target File_Type [copy to here]
scope: # Copy to the target File_Type in this scope [if not specified the default is sample]
collect_results: # Will copy (symbolic link) selected files to a Results folder
sample: #
File_Type: # list of sample scope File_Type separated by comma to copy ['fastq.F,fastq.R']
project: #
File_Type: # list of project scope File_Type separated by comma to copy ['fastq.F,fastq.R']
qsub_params: # Parameters for qsub [number of cpus or memory to reserve etc ]
STR:
redirects: # Parameters to pass directly to the command
STR:
Fillout_Generic
- Authors
Menachem Sklarz
- Affiliation
Bioinformatics core facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
Description
This module enables executing any type of bash command, including pipes and multiple steps.
File and directory names are embedded in the script by describing the file or directory in a {{}} block, as follows:
1. File names:
Include 4 colon-separated fields: (a) scope, (b) slot, (c) separator and (d) base.
For example: {{sample:fastq.F:,:merge1}} is replaced with sample fastq.F files from merge1 instance, seperated by commas (only for project scope scripts, of course).
Leave fields empty if you do not want to pass a value, e.g. {{sample:fastq.F}} is replaced with the sample fastq.F file.
If the partition option is used: (a) can be set to part.
2. Sample and project names:
You can include the sample or project names in the script by leaving out the file type field. e.g. {{sample}} will be replaced by the sample name.
To get a list of sample names, set the separator field to the separator of your choice, e.g. {{sample::,}} will be replaced with a comma-separated list of sample names.
If the partition option is used: (a) can be set to part and if (b) is not set it will use the part number for numeric partition or line in the file used in the partition option. In this case (d) can be set to “clean” , “basename” or “clean,basename” to use only the basename of part. If clean is used will replace spaces with underscore and strip whitespaces
3. Directories
You can include two directories in your command:
Dir descriptor |
Result |
|---|---|
|
Returns the base directory for the step. |
|
Returns the active directory of the script. For project-scope scripts, this is identical to |
Tip
You can obtain the base_dir or dir values for a base step, by including the name of the base in the 4th colon separated position, just as you’d do for the file slots. e.g. {{base_dir:::merge1}} will return the base_dir for step merge1 and {{dir:::merge1}} will return the dir for the current sample for step merge1.
3. Outputs
Will be replaced with the filename specified in the named output. e.g. {{o:fasta.nucl}} will be replced according to the specifications in the output block named fasta.nucl.
Each output block must contain 2 fields: scope and string. The string contains a string describing the file to be stored in the equivalent slot. In the example above, there must be a block called fasta.nucl in the output block which can be defined as shown in the example in section Lines for parameter file below.
3. Examples
The following examples cover most of the options:
File descriptor |
Result |
|---|---|
|
The |
|
The |
|
A comma-separated list of the |
|
The project name |
|
The sample name |
|
A comma-separated list of sample names |
|
A comma-separated list of the |
|
If the partition option is used, returns the part number for numeric partition or line in the file used in the partition option. If basename is added it will use only the basename of part. If clean is used will replace spaces with underscore and strip whitespaces |
|
If the partition option is used, returns the file type fastq.F in the current part |
Tip
For a colon separate list of sample names or files, use the word ‘colon’ in the separator slot.
Note
The separator field is ignored for project-scope slots.
Attention
If a sample-scope slot is used, in the inputs or the outputs, the scripts will be sample-scope scripts. Otherwise, one project-scope script will be produced. To override this behaviour, set scope to project.
However, you cannot set scope to project if there are sample-scope fields defined.
Requires:
Customizable
Output:
Customizable
Parameters that can be set
Parameter |
Values |
Comments |
|---|---|---|
output |
A block including ‘scope’ and ‘string’ definining the script outputs |
|
scope |
|
The scope of the resulting scripts. You cannot set scope to project if there are sample-scope fields defined. |
partition |
|
Can split the analysis to number of parts or by lines in a file. |
del_partition |
If in Previous steps the |
Lines for parameter file
Demonstration of embedding various files and titles in a script file:
pipe_gen_3:
module: Fillout_Generic
base: pipe_gen_2
script_path: |
project: {{project}}
fasta.nucl in project: {{project:fasta.nucl}}
fasta.nucl in project from base merge1: {{project:fasta.nucl::merge1}}
sample names: {{sample::,}}
fastq.F in sample: {{sample:fastq.F}}
fastq.F in sample from base merge1: {{sample:fastq.F::merge1}}
output:fasta.nucl: {{o:fasta.nucl}}
output:
fasta.nucl:
scope: project
string: "{{base_dir}}{{project}}_new_pipegen3.fasta"
Comments