Variant calling
- Author
Menachem Sklarz
- Affiliation
Bioinformatics Core Facility
- Organization
National Institute of Biotechnology in the Negev, Ben Gurion University.
This workflow performs a basic variant-calling analysis.
Steps:
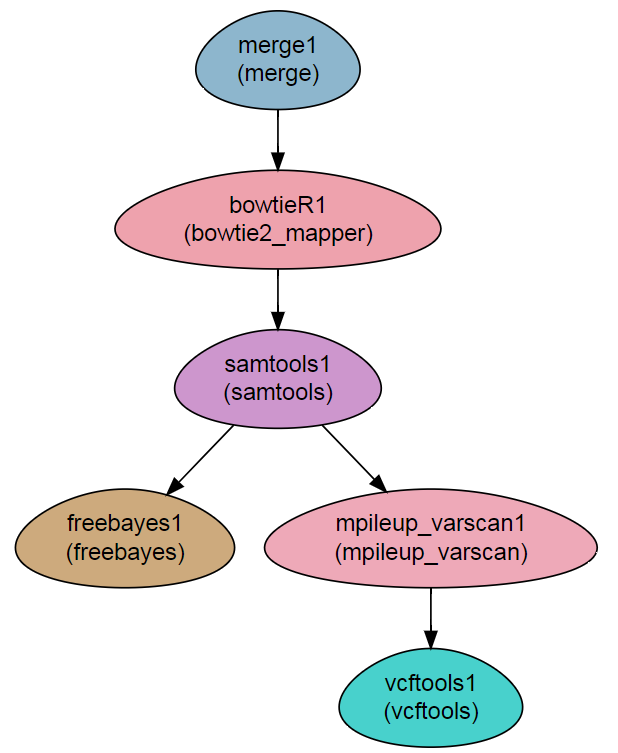
Merging the fastq sequences into a single file per sample (
merge)Mapping to a reference genome with bowtie2 (
bowtie2module)Sorting, filtering and conversion to BAM with samtools (
samtoolsmodule)- Variant calling with two programs:
freebayes (
freebayesmodule)mpileup and varscan (
mpileup_varscanmodule)
Analysis of the resulting VCF file with VCFtools (
vcftoolsmodule).
Workflow Schema
Requires
fastq files, either paired-end or single-end.
Programs required
Example of Sample File
Title ChIP_project
#SampleID Type Path lane
Sample1 Forward /path/to/Sample1_F1.fastq.gz 1
Sample1 Forward /path/to/Sample1_F2.fastq.gz 2
Sample1 Reverse /path/to/Sample1_R1.fastq.gz 1
Sample1 Reverse /path/to/Sample1_R2.fastq.gz 2
Sample2 Forward /path/to/Sample2_F1.fastq.gz 1
Sample2 Reverse /path/to/Sample2_R1.fastq.gz 1
Sample2 Forward /path/to/Sample2_F2.fastq.gz 2
Sample2 Reverse /path/to/Sample2_R2.fastq.gz 2
Download
The workflow file is available here